Theme: Pharmaceutical analysis of phenothiazine and benzodiazepine derivatives as drug substances: synthesis, properties, analysis, storage, action and use.
Phenothiazines
Phenothiazine is an organic compound that occurs in various antipsychotic and antihistaminic drugs. It has the formula S(C6H4)2NH. This is tricyclic compound (is a fused heterocyclic system, which consists of six-membered heterocycle (thiazine) and two benzene rings). The compounds are related to the thiazine-class of heterocyclic compounds.


10H-phenothiazine
Derivatives of the parent compound find wide use as drugs.

Structure of phenothiazine derivatives
Medicinal substances, phenothiazine derivatives, depending on the nature of the radical at position 10, have various pharmacological actions.
For example, 10-alkyl derivatives of phenothiazine (Levomepromazine hydrochloride, Promazine hydrochloride, Chlorpromazine hydrochloride, Perphenazine, Trifluoperazine hydrochloride* and Promethazine hydrochloride**) are used as antipsychotic (neuroleptic)* and antihistamine agents**, and 10-acyl derivatives (Moracizine, Ethacizine (antiarrhythmic agents)) are effective in the treatment of cardiovascular system.
Phenothiazine derivatives have the ability to penetrate into the body through the respiratory tract, skin and mucous membranes. Thus they cause allergic reactions. Therefore, is necessary strictly adhere to safety in the work, excluding the possibility of hit this group of drugs on exposed skin and mucous membranes. After finishing of the work the hands should be washed cold water, slightly acidified (no soap) (to prevent the formation on the skin bases of appropriate phenothiazine derivatives).
Phenothiazine-derived drugs
The phenothiazine structure occurs in various neuroleptic drugs, e.g. chlorpromazine, and antihistaminic drugs, e.g. promethazine. The term “phenothiazines” describes the largest of the five main classes of neuroleptic antipsychotic drugs. These drugs have antipsychotic and, often, antiemetic properties, although they may also cause severe side effects such as extrapyramidal symptoms (including akathisia and tardive dyskinesia), hyperprolactinaemia, and the rare but potentially fatal neuroleptic malignant syndrome as well as substantial weight gain.
Phenothiazines are used as inodilators in congestive heart failure, acting upon the type I calcium/calmodulin dependent phosphodiesterase.
Phenothiazine antipsychotics are classified into three groups that differ with respect to the substituent oitrogen: the aliphatic compounds (bearing acyclic groups), the “piperidines” (bearing piperidine-derived groups), and the piperazine (bearing piperazine-derived substituents).
|
Group |
Example |
Sedative |
||
|
|
|
|
|
|
|
moderate |
Chlorpromazine (marketed as Thorazine, Chlor-PZ, Klorazine, Promachlor, Promapar, Sonazine, Chlorprom, Chlor-Promanyl, Largactil) |
strong |
moderate |
|
|
Promazine (trade name Sparine) |
moderate |
moderate |
||
|
Triflupromazine (trade names Stelazine, Clinazine, Novaflurazine, Pentazine, Terfluzine, Triflurin, Vesprin) |
strong |
moderate/strong |
||
|
Levomepromazine in |
extremely strong |
low |
||
|
strong |
Mesoridazine (trade name Serentil) |
strong |
weak |
|
|
Thioridazine (trade names Mellaril, Novoridazine, Thioril) |
strong |
weak |
||
|
weak |
Fluphenazine (trade names Prolixin, Permitil, Modecate, Moditen) |
weak/moderate |
strong |
|
|
Perphenazine (sold as Trilafon, Etrafon, Triavil, Phenazine) |
weak/moderate |
strong |
||
|
Prochlorperazine (trade names Compazine, Stemetil) |
|
|
||
|
Trifluoperazine (trade name Stelazine) |
moderate |
strong |
Tradenames for phenothiazine
Like many commercially significant compounds, phenothiazine has numerous trade names including AFI-Tiazin; Agrazine; Antiverm; Biverm; Dibenzothiazine; Orimon; Lethelmin; Souframine; Nemazene; Vermitin; Padophene; Fenoverm; Fentiazine; Contaverm; Fenothiazine; Phenovarm; Ieeno; ENT 38; Helmetina; Helmetine, Penthazine; XL-50; Wurm-thional; Phenegic; Phenovis; Phenoxur; Reconox.
Chlorpromazine Tablets
Chlorpromazini tabulettae obductae
![]()
One chlorpromazine tablet consists of
Chlorpromazine Hydrochloride
Chlorpromazini hydrochloridum
C17H19ClN2S, HCl
355.3
DEFINITION
Chlorpromazine Tablets contain Chlorpromazine Hydrochloride (3-(2-Chloro-10H–phenothiazin-10-yl)-N,N–dimethylpropan-1-amine hydrochloride). They are coated.
The tablets comply with the requirements stated under Tablets and with the following requirements.
Content of chlorpromazine hydrochloride, C17H19ClN2S, HCl.
92.5 to 107.5% of the stated amount.
CHARACTERS
Chlorpromazine tablets are coated and with a pink colour.
IDENTIFICATION
To 4 powdered tablets add 20 ml of water, gently shake and filtrate. The received filtrate gives reactions of identification:
Non-Pharmacopoeial reactions:
– with oxidants (concentrated H2SO4 (A), HNO3 (B), bromine water (C) and others):
A. To 1 ml of filtrate add 0,1 ml of 0,1 % methylene blue solution and 2 ml of concentrated sulphuric acid. A purple colour is produced.
B. To 1 ml of filtrate add 2 drops of concentrated nitric acid. A red colour and opalescence is produced. After that add 2-3 drops of concentrated nitric acid; solution becomes a transparent and colorless.
C. (more specific reaction). To 1 ml of filtrate add 1 ml of bromine water and heat to boiling. A clear pale-pink colour is produced.
The probable structure of the obtained product:
D. To 5 ml of filtrate add 0,5 ml of dilute sodium hydroxide; white precipitate is formed. After 5 minutes filtered through dense paper filter. The filtrate gives reaction (a) of chlorides.
Chlorides:
A. Reaction with silver nitrate solution in the niric-acid medium
2 ml of filtrate acidify with dilute nitric acid and add 0.4 ml of silver nitrate solution. Shake and allow to stand. A curdled, white precipitate is formed. Centrifuge and wash the precipitate with three quantities, each of 1 ml, of water. Carry out this operation rapidly in subdued light, disregarding the fact that the supernatant solution may not become perfectly clear. Suspend the precipitate in 2 ml of water R and add 1.5 ml of ammonia. The precipitate dissolves easily with the possible exception of a few large particles which dissolve slowly.
Cl– + Ag+ → AgCl↓
white curdled precipitate
AgCl↓ + 2NH4OH → [Ag(NH3)2]Cl + 2H2O
ASSAY
(Non-Pharmacopoeial method). Alkalimetry, direct titration
5 tablets dissolve in 25 ml of water and add
1 ml of
Em (C17H20Cl2N2S)= М. m.
STORAGE
In an airtight container , protected from light.
Action and use
Antipsychotic; antiemetic.
Ethacizine
CAS registry number (Chemical Abstracts Service)
0033414-33-4
Chemical Formula
C22-H27-N3-O3-S
Molecular Weight
413
Therapeutic Category
Antiarrhythmic agent
Chemical Names
[10-[3-(Diethylamino)-1-oxopropyl]-10H–phenothiazin-2-yl]carbamic acid ethyl ester
10-(N,N–Diethyl–beta–alanyl)-phenothiazine-2-carbamic acid ethyl ester
10-[2-(Diethylamino)acetyl]phenothiazin-2-ylcarbamidsäureethylester (IUPAC)
Carbamic acid, (10-(3-(diethylamino)-1-oxopropyl)-10H–phenothiazin-2-yl)-, ethyl ester
Ethyl (10-(3-(diethylamino)-1-oxopropyl)-10H–phenothiazin-2-yl)carbamate
Ethyl 10-[3-(diethylamino)propionyl]phenothiazine-2-carbamate
Phenothiazine-2-carbamic acid, 10-(N,N–diethyl–beta–alanyl)-, ethyl ester (8CI)
Foreign Name
· Ethacizin (German)
Generic Names
· BRN 4827533 (IS)
· DAAE (IS)
· Etacizin (IS)
· ETHA (IS)
· Ethacyzin (IS)
· Ethmozine DAA (IS)
· Etmozine DAA (IS)
· EZ 55 (IS)
· NIK 244 (IS)
Brand Name
· Etacizins
Olainfarm,
- Synthesis of Ethacizine
· Ethacizine (CAS NO.: 33414-33-4), with its systematic name of Phenothiazine-2-carbamic acid, 10-(N,N-diethyl-beta-alanyl)-, ethyl ester (8CI), could be produced through many synthetic methods.
Following is one of the synthesis routes: 10-(3-Chloropropionyl)-2-(ethoxycarbonylamino)phenothiazine (I) reacts with diethylamine in the presence of refluxing toluene to produce Ethacizine.
Etacizins may be available in the countries listed below.
Ingredient matches for Etacizins
Ethacizine is reported as an ingredient of Etacizins in the following countries:
·
Ethacizin: pharmacologic properties and prospects for clinical application.
Abstract
The study of the regularities between the chemical structure and pharmacologic action of phenathiazine dialkylaminoacyl derivatives led to the identification and investigation of a new drug called ethacizine-phenothiazin-2-carbethoxyamino-10 (beta-diethylamino-propionyl) hydrochloride. Ethacizine exceeds its structural analogue ethmozin by two times in terms of intensity and by 4-5 times in terms of the duration of antiarrhythmic effect. Ethacizine has marked antianginal properties. It shows a prolonged inhibitory effect on the average elevation of the ST interval at multiple leads of the epicardial electrogram during coronary occlusion, increases the threshold of myocardial ischemia development, and reduces the size of experimental infarction. The combination of potent antiarrhythmic activity, already confirmed by clinical observations, with antianginal properties and a capacity to limit the size of infarction makes in possible to consider ethacizine a promising means for treating coronary heart disease.
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Levomepromazine Hydrochloride
Levomepromazine Hydrochloride
General Notices
(Ph Eur monograph 0505)
C19H24N2OS,HClээ364.9ээ4185-80-2
DEFINITION
Levomepromazine hydrochloride contains not less than 98.5 per cent and not more than the equivalent of 101.0 per cent of (2R)-3-(2-methoxy-10H-phenothiazin-10-yl)-N,N,2– trimethylpropan-1-amine hydrochloride, calculated with reference to the dried substance.
CHARACTERS
A white or very slightly yellow, crystalline powder, slightly hygroscopic, freely soluble in water
and in alcohol. It deteriorates when exposed to air and light. It exists in two forms, one melting
at about
IDENTIFICATION
эA. Prepare the solution protected from bright light and carry out the measurements immediately. Dissolve 50.0 mg in water R and dilute to 500.0 ml with the same solvent. Dilute 10.0 ml of this solution to 100.0 ml with water R. Examined between 230 nm and 340 nm (2.2.25), the solution shows two absorption maxima, at 250 nm and 302 nm. The specific absorbance at the maximum at 250 nm is 640 to 700.
эB. It complies with the identification test for phenothiazines by thin-layer chromatography (2.3.3): use levomepromazine hydrochloride CRS to prepare the reference solution.
эC. Introduce
эD. It gives reaction (b) of chlorides (2.3.1).
Chlorides
B. Introduce into a test-tube a quantity of the substance to be examined equivalent to about 15 mg of chloride (Cl–) or the prescribed quantity. Add
a filter-paper strip impregnated with 0.1 ml of diphenylcarbazide solution R over the opening of the test-tube. The paper turns violet-red. The impregnated paper must not come into contact with the potassium dichromate.
TESTS
Solution S
Dissolve
Acidity or alkalinity
To 10 ml of solution S add 0.1 ml of bromocresol green solution R. Not more than 0.5 ml of
Specific optical rotation (2.2.7)
+ 9.5 to + 11.5, determined on solution S and calculated with reference to the dried substance.
Related substances
Carry out the test protected from bright light. Examine by thin-layer chromatography (2.2.27), using silica gel GF254 R as the coating substance.
Test solution Dissolve
Reference solution Dilute 0.5 ml of the test solution to 100 ml with a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R.
Apply separately to the plate 10 µl of each solution. Develop over a path of
Loss on drying (2.2.32)
Not more than 1.0 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
ASSAY
Dissolve
STORAGE
Store in an airtight container, protected from light.
Ph Eur
Action and use
Dopamine receptor antagonist; neuroleptic. (ВР 2009)
Antipsychotic.(ВР 2007)
Preparation
Levomepromazine Injection
Ph Eur
Moracizine
|
Moracizine |
|
|
|
|
|
ethyl N-[10-(3-morpholin-4-ylpropanoyl)phenothiazin-2-yl]carbamate |
|
|
Identifiers |
|
|
Jmol-3D images |
|
|
Properties |
|
|
C22H25N3O4S |
|
|
427.5166 |
|
|
|
|
|
|
|

Moracizine is an antiarrhythmic. It is used for the prophylaxis and treatment of serious and life-threatening ventricular arrhythmias, in the
British National Formulary 59
· Cardiovascular system drug stubs
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Perphenazine
Perphenazine
General Notices
(Ph Eur monograph 0629)
C21H26ClN3OSээ404.0ээ58-39-9
DEFINITION
Perphenazine contains not less than 99.0 per cent and not more than the equivalent of 101.0 per cent of 2-[4-[3-(2-chlorophenothiazin-10-yl)propyl]piperazin-1-yl]ethanol, calculated with reference to the dried substance.
CHARACTERS
A white or yellowish-white, crystalline powder, practically insoluble in water, freely soluble in methylene chloride, soluble in alcohol. It dissolves in dilute solutions of hydrochloric acid.
IDENTIFICATION
First identificationэA, C
Second identificationэA, B, D.
эA. Melting point (2.2.14):
эB. Dissolve 10 mg in methanol R and dilute to 100 ml with the same solvent. Dilute 10 ml of the solution to 100 ml with methanol R. Examined between 230 nm and 350 nm (2.2.25), the solution shows two absorption maxima, at 257 nm and 313 nm. The ratio of the absorbance measured at the maximum at 313 nm to that measured at the maximum at 257 nm is 0.120 to 0.128.
э
C. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with perphenazine CRS . Examine the substances prepared as discs.
D. Examine by thin-layer chromatography (2.2.27), using kieselguhr G R as the coating substance. Impregnate the plate by placing it in a closed tank containing the necessary quantity of the impregnation mixture containing 2.5 per cent V/V of phenoxyethanol R and 7.5 per cent V/V of formamide R in acetone R so that the plate dips about
Test solution Dissolve 20 mg of the substance to be examined in chloroform R and dilute to 10 ml with the same solvent.
Reference solution Dissolve 20 mg of perphenazine CRS in chloroform R and dilute to 10 ml with the same solvent.
Apply separately to the plate 2 µl of each solution. Develop in the dark over a path of
TESTS
Appearance of solution
Dissolve
Related substances
Examine by thin-layer chromatography (2.2.27), using silica gel GF254 R as the coating substance. Prepare the solutions immediately before use.
Test solution Dissolve
Reference solution Dilute 0.5 ml of the test solution to 100 ml with methanol R.
Apply separately to the plate 10 µl of each solution. Develop over a path of
Loss on drying (2.2.32)
Not more than 0.5 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
ASSAY
Dissolve
1 ml of
STORAGE
Store protected from light.
Ph Eur
Action and use
Dopamine receptor antagonist; neuroleptic. (ВР 2009)
Antipsychotic; antiemetic. (ВР 2007)
Preparation
Perphenazine Tablets
Ph Eur
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Promazine Hydrochloride
Promazine Hydrochloride
General Notices
(Ph Eur monograph 1365)
C17H20N2S,HCl 320.9 53-60-1
DEFINITION
Promazine hydrochloride contains not less than 99.0 per cent and not more than the equivalent of 101.0 per cent of 3-(10H-phenothiazin-10-yl)-N,N-dimethylpropan-1-amine hydrochloride, calculated with reference to the dried substance.
CHARACTERS
A white or almost white, crystalline powder, slightly hygroscopic, very soluble in water, in alcohol and in methylene chloride.
It melts at about
IDENTIFICATION
First identification A, B, D.
Second identification B, C, D.
A. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with promazine hydrochloride CRS.
B. It complies with the identification test for phenothiazines by thin-layer chromatography (2.3.3). Use promazine hydrochloride CRS to prepare the reference solution.
C. Dissolve about 5 mg in 2 ml of sulphuric acid R and allow to stand for 5 min. An orange colour is produced.
D. It gives reaction (b) of chlorides (2.3.1).
Chlorides
B. Introduce into a test-tube a quantity of the substance to be examined equivalent to about 15 mg of chloride (Cl–) or the prescribed quantity. Add
a filter-paper strip impregnated with 0.1 ml of diphenylcarbazide solution R over the opening of the test-tube. The paper turns violet-red. The impregnated paper must not come into contact with the potassium dichromate.
TESTS
pH (2.2.3)
Dissolve
Related substances
Carry out the test protected from bright light. Prepare the solutions immediately before use.
Examine by thin layer chromatography (2.2.27), using a TLC silica gel F254plate R.
Test solution Dissolve
Reference solution (a) Dilute 1 ml of the test solution to 200 ml with a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R.
Reference solution (b) Dissolve 10 mg of chlorprothixene hydrochloride CRS in a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R, add 1 ml of the test solution and dilute to 10 ml with the same mixture of solvents.
Apply to the plate 10 µl of each solution. Develop over a path of
Loss on drying (2.2.32)
Not more than 0.5 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
ASSAY
Dissolve
1 ml of
STORAGE
Store protected from light.
IMPURITIES
A. 3-(10H-phenothiazin-10-yl)-N,N-dimethylpropan-1-amine S-oxide (promazine sulphoxide).
Ph Eur
Action and use
Antipsychotic. (ВР 2007)
Dopamine receptor antagonist; neuroleptic. (ВР 2009)
Preparations
Promazine Injection
Promazine Oral Suspension
Promazine Tablets
Ph Eur
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Promethazine Hydrochloride
Promethazine Hydrochloride
General Notices
(Ph Eur monograph 0524)
C17H20N2S,HCl 320.9 58-33-3
DEFINITION
Promethazine hydrochloride contains not less than 99.0 per cent and not more than the equivalent of 101.0 per cent of (2RS)-N,N-dimethyl-1-(10H-phenothiazin-10-yl)propan-2-amine hydrochloride, calculated with reference to the dried substance.
CHARACTERS
A white or faintly yellowish, crystalline powder, very soluble in water, freely soluble in alcohol and in methylene chloride.
It melts at about
IDENTIFICATION
First identification A, B, D.
Second identification B, C, D.
A. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with promethazine hydrochloride CRS.
B. It complies with the identification test for phenothiazines by thin-layer chromatography (2.3.3).
C. Dissolve
D. It gives reaction (b) of chlorides (2.3.1).
Chlorides
B. Introduce into a test-tube a quantity of the substance to be examined equivalent to about 15 mg of chloride (Cl–) or the prescribed quantity. Add
a filter-paper strip impregnated with 0.1 ml of diphenylcarbazide solution R over the opening of the test-tube. The paper turns violet-red. The impregnated paper must not come into contact with the potassium dichromate.
TESTS
pH (2.2.3)
Dissolve
Related substances
Carry out the test protected from bright light. Prepare the solutions immediately before use. Examine by thin-layer chromatography (2.2.27), using a suitable silica gel as the coating substance.
Test solution Dissolve
Reference solution (a) Dissolve 20 mg of isopromethazine hydrochloride CRS in a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R and dilute to 100 ml with the same mixture of solvents.
Reference solution (b) Dilute 0.5 ml of the test solution to 100 ml with a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R.
Reference solution (c) Dilute 0.2 ml of the test solution to 100 ml with a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R.
Apply separately to the plate 10 µl of the test solution and 10 µl of each reference solution. Develop in an unsaturated tank over a path of
In the chromatogram obtained with the test solution: any spot corresponding to isopromethazine hydrochloride is not more intense that the spot in the chromatogram obtained with reference solution (a) (1 per cent); any spot, apart from the principal spot and the spot corresponding to isopromethazine hydrochloride, is not more intense than the spot in the chromatogram obtained with reference solution (b) (0.5 per cent) and at most three such spots are more intense than the spot in the chromatogram obtained with reference solution (c) (0.2 per cent).
Heavy metals (2.4.8)
Dissolve
Loss on drying (2.2.32)
Not more than 0.5 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
ASSAY
Dissolve
1 ml of
STORAGE
Store protected from light.
IMPURITIES
A. phenothiazine,
B. (2RS)-N,N-dimethyl-2-(10H-phenothiazin-10-yl)propan-1-amine (isopromethazine),
C. R = H, X = S: (2RS)-N–methyl-1-(10H–phenothiazin-10-yl)propan-2-amine,
D. R = CH3, X = SO: (2RS)-N,N–dimethyl-1-(10H–phenothiazin-10-yl)propan-2-amine S–oxide.
Ph Eur
Action and use
Histamine H1-receptor antagonist; anti-emetic.
Preparations
Promethazine Injection
Promethazine Oral Solution
Promethazine Hydrochloride Tablets
Ph Eur
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Trifluoperazine Hydrochloride
Trifluoperazine Hydrochloride
General Notices
(Ph Eur monograph 0059)
C21H24F3N3S,2HClээ480.4ээ 440-17-5
DEFINITION
Trifluoperazine hydrochloride contains not less than 99.0 per cent and not more than the
equivalent of 101.0 per cent of 10-[3-(4-methylpiperazin-1-yl)propyl]-2-(trifluoromethyl)-10H–phenothiazine dihydrochloride, calculated with reference to the dried substance.
CHARACTERS
A white to pale yellow, crystalline powder, hygroscopic, freely soluble in water, soluble in
alcohol.
It melts at about
IDENTIFICATION
эA. Protect the solutions from bright light and measure the absorbances immediately. Dissolve 50 mg in
Э
B. It complies with the identification test for phenothiazines by thin-layer chromatography (2.3.3) : use trifluoperazine hydrochloride CRS to prepare the reference solution.
эC. Place
эD. Dissolve about 0.5 mg in 1 ml of water R , add 0.1 ml of bromine water R and shake for about 1 min. Add dropwise 1 ml of sulphuric acid R with constant, vigorous agitation. A red colour develops.
эE. Dissolve about 50 mg in 5 ml of water R and add 2 ml of nitric acid R. A dark-red colour develops which turns to pale yellow. The solution gives reaction (a) of chlorides (2.3.1).
Chlorides
A. Dissolve in 2 ml of water R a quantity of the substance to be examined equivalent to about 2 mg of chloride (Cl–) or use 2 ml of the prescribed solution. Acidify with dilute nitric acid R and add 0.4 ml of silver nitrate solution R1. Shake and allow to stand. A curdled, white precipitate is formed. Centrifuge and wash the precipitate with three quantities, each of 1 ml, of water R. Carry out this operation rapidly in subdued light, disregarding the fact that the supernatant solution may not become perfectly clear. Suspend the precipitate in 2 ml of water R and add 1.5 ml of ammonia R. The precipitate dissolves easily with the possible exception of a few large particles which dissolve slowly.
TESTS
pH (2.2.3)
Dissolve
Related substances
Carry out the test protected from bright light.
Examine by thin-layer chromatography (2.2.27), using a TLC silica gel GF254plate R.
Test solution Dissolve
Reference solution Dilute 1 ml of the test solution to 200 ml with a mixture of 5 volumes of diethylamine R and 95 volumes of methanol R.
Apply to the plate 10 µl of each solution. Develop over a path of
Loss on drying (2.2.32)
Not more than 1.5 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
ASSAY
Dissolve
1 ml of
STORAGE
Store in an airtight container , protected from light.
Ph Eur
Action and use
Dopamine receptor antagonist; neuroleptic. (ВР 2009)
Antipsychotic.(ВР 2007)
Preparation
Trifluoperazine Tablets
Ph Eur
Benzodiazepines
A benzodiazepine (sometimes colloquially “benzo“; often abbreviated “BZD“) is a psychoactive drug whose core chemical structure is the fusion of a benzene ring and a diazepine ring (1,4-diazepine) (seven-membered heterocycle).
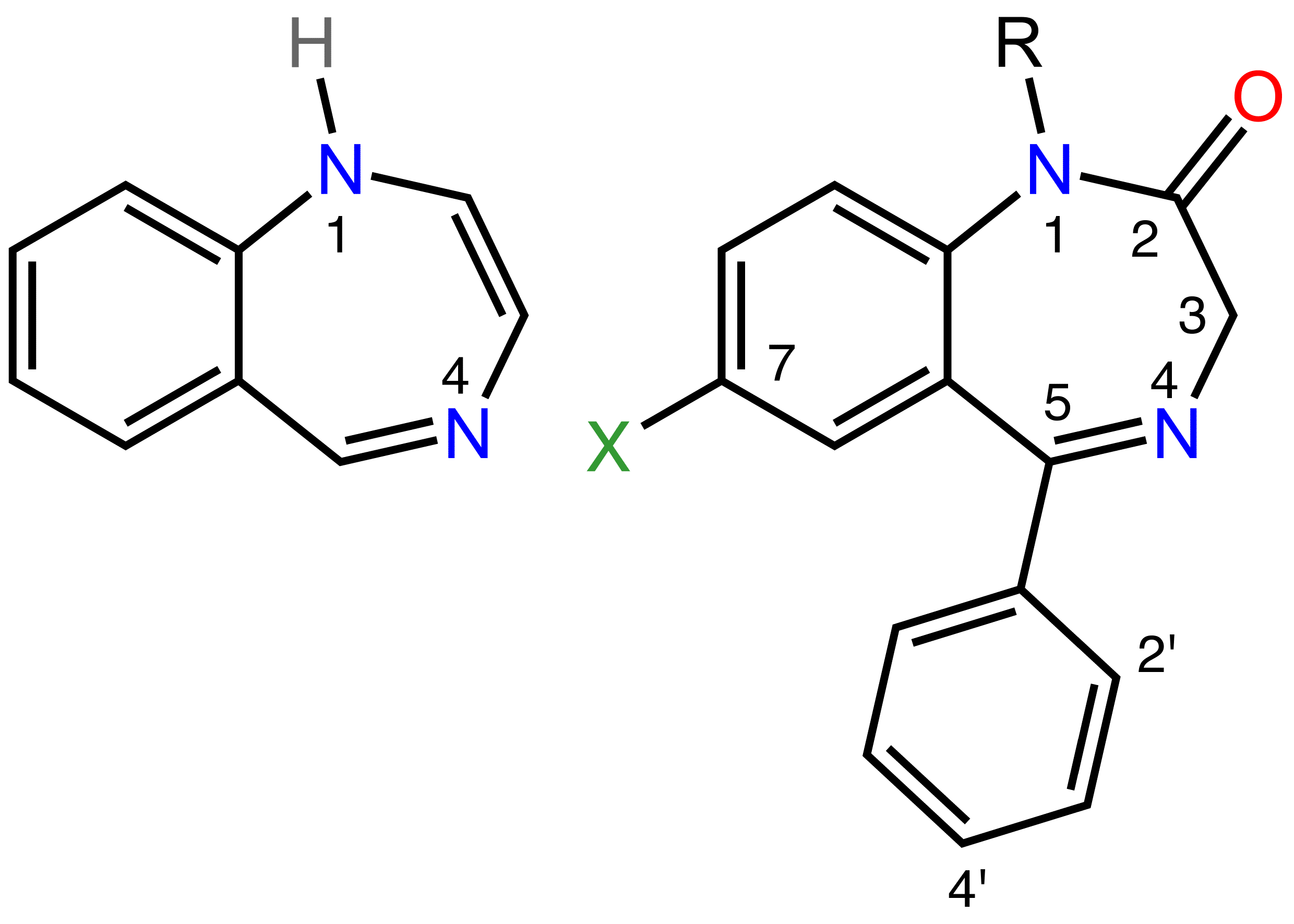
Left: The 1,4-benzodiazepine ring system.
Right: 5-phenyl-1H-benzo[e][1,4]diazepin-2(3H)-one forms the skeleton of many of the most common benzodiazepine pharmaceuticals, such as Oxazepam (7-chloro-3-hydroxy-5-phenyl substituted) (use as anxiolytic) and Nitrazepam (7-nitro-5-phenyl substituted) (use as hypnotic).
A benzodiazepine is a psychoactive drug. Benzodiazepines have a sedative, hypnotic (sleep-inducing), anxiolytic (anti-anxiety), anticonvulsant, muscle relaxant and amnesic action. These properties make benzodiazepines useful in treating anxiety, insomnia, agitation, seizures, muscle spasms, alcohol withdrawal and as a premedication for medical or dental procedures.
Benzodiazepines enhance the effect of the neurotransmitter gamma-aminobutyric acid (GABA), which results in sedative, hypnotic (sleep-inducing), anxiolytic (anti-anxiety), anticonvulsant, muscle relaxant and amnesic action. These properties make benzodiazepines useful in treating anxiety, insomnia, agitation, seizures, muscle spasms, alcohol withdrawal and as a premedication for medical or dental procedures. Benzodiazepines are categorized as either short-, intermediate- or long-acting. Short- and intermediate-acting benzodiazepines are preferred for the treatment of insomnia; longer-acting benzodiazepines are recommended for the treatment of anxiety.
In general, benzodiazepines are safe and effective in the short term, although cognitive impairments and paradoxical effects such as aggression or behavioral disinhibition occasionally occur. Long-term use is controversial due to concerns about adverse psychological and physical effects, increased questioning of effectiveness and because benzodiazepines are prone to cause tolerance, physical dependence, and, upon cessation of use after long term use, a withdrawal syndrome. Due to adverse effects associated with the long-term use of benzodiazepines, withdrawal from benzodiazepines, in general, leads to improved physical and mental health. The elderly are at an increased risk of suffering from both short- and long-term adverse effects.

Structure of benzodiazepine derivatives
Benzodiazepines possess sedative, hypnotic, anxiolytic, anticonvulsant, muscle relaxant, and amnesic actions, which are useful in a variety of indications such as alcohol dependence, seizures, anxiety, panic, agitation and insomnia. Most are administered orally; however, they can also be given intravenously, intramuscularly or rectally. In general, benzodiazepines are well-tolerated and are safe and effective drugs in the short term for a wide range of conditions.
Drugs from this group have started to apply with the beginning of 60th years ХХ century. Now in world practice are applied about 20 preparations from group 1,4-benzodiazepine. The modern tranquilizers possessing sedative effect at the minimum influence on impellent and cogitative functions concern them. Unlike neuroleptics do not show antipsychotic activity. They possess antidisturbing, sedative-hypnotic, somnolent (hypnagogue), anticonvulsant action.
In chemical structure of molecules of these preparations contains phenylic radical in position 5 and they are derivatives of 5-phenyl-3H-1,4-benzodiazepine (Chlordiasepoxidum, Mezapamum) and 1,2-dihydro-3Н-1,4- benzodiazepine-2-one (Diazepamum, Oxazepam, Nitrazepamum, Phenazepamum, etc.).
Benzodiazepine drugs are substituted 1,4-benzodiazepines, although the chemical term can refer to many other compounds that do not have useful pharmacological properties. Different benzodiazepine drugs have different side groups attached to this central structure. The different side groups affect the binding of the molecule to the GABAA receptor and so modulate the pharmacological properties. Many of the pharmacologically active “classical” benzodiazepine drugs contain the 5-phenyl-1H-benzo[e][1,4]diazepin-2(3H)-one substructure (see figure to the right).
Mechanism of action

![]()
Schematic diagram of the (α1)2(β2)2(γ2) GABAA receptor complex that depicts the five-protein subunits that form the receptor, the chloride (Cl–) ion channel pore at the center, the two GABA active binding sites at the α1 and β2 interfaces and the benzodiazepine (BZD) allosteric binding site at the α1 and γ2 interface.
Benzodiazepines work by increasing the efficiency of a natural brain chemical, GABA, to decrease the excitability of neurons. This reduces the communication betweeeurons and, therefore, has a calming effect on many of the functions of the brain.
GABA controls the excitability of neurons by binding to the GABAA receptor.[112] The GABAA receptor is a protein complex located in the synapses of neurons. All GABAA receptors contain an ion channel that conducts chloride ions across neuronal cell membranes and two binding sites for the neurotransmitter gamma-aminobutyric acid (GABA), while a subset of GABAA receptor complexes also contain a single binding site for benzodiazepines. Binding of benzodiazepines to this receptor complex promotes binding of GABA, which in turn increases the conduction of chloride ions across the neuronal cell membrane. This increased conductance raises the membrane potential of the neuron, resulting in inhibition of neuronal firing. In addition, different GABAA receptor subtypes have varying distributions within different regions of the brain and, therefore, control distinct neuronal circuits. Hence, activation of different GABAA receptor subtypes by benzodiazepines may result in distinct pharmacological actions.[118] In terms of the mechanism of action of benzodiazepines, their similarities are too great to separate them into individual categories such as anxiolytic or hypnotic. For example, a hypnotic administered in low doses will produce anxiety-relieving effects, whereas a benzodiazepine marketed as an anti-anxiety drug will at higher doses induce sleep.[119]
The subset of GABAA receptors that also bind benzodiazepines are referred to as benzodiazepine receptors (BzR). The GABAA receptor is a heteromer composed of five subunits, the most common ones being two αs, two βs, and one γ (α2β2γ). For each subunit, many subtypes exist (α1-6, β1-3, and γ1-3). GABAA receptors that are made up of different combinations of subunit subtypes have different properties, different distributions in the brain and different activities relative to pharmacological and clinical effects.[120] Benzodiazepines bind at the interface of the α and γ subunits on the GABAA receptor. Binding also requires that alpha subunits contain a histidine amino acid residue, (i.e., α1, α2, α3, and α5 containing GABAA receptors). For this reason, benzodiazepines show no affinity for GABAA receptors containing α4 and α6 subunits with an arginine instead of a histidine residue.[121]
Once bound to the benzodiazepine receptor, the benzodiazepine ligand locks the benzodiazepine receptor into a conformation in which it has a greater affinity for the GABA neurotransmitter. This increases the frequency of the opening of the associated chloride ion channel and hyperpolarizes the membrane of the associated neuron. The inhibitory effect of the available GABA is potentiated, leading to sedatory and anxiolytic effects. Furthermore, different benzodiazepines can have different affinities for BzRs made up of different collection of subunits. For instance, those with high activity at the α1 are associated with stronger hypnotic effects, whereas those with higher affinity for GABAA receptors containing α2 and/or α3 subunits have good anti-anxiety activity.[122]
The benzodiazepine class of drugs also interact with peripheral benzodiazepine receptors. Peripheral benzodiazepine receptors are present in peripheral nervous system tissues, glial cells, and to a lesser extent the central nervous system.[123] These peripheral receptors are not structurally related nor coupled to GABAA receptors. They modulate the immune system and are involved in the body response to injury.[113][124] Benzodiazepines also function as weak adenosine reuptake inhibitors. It has been suggested that some of their anticonvulsant, anxiolytic and muscle relaxant effects may be in part mediated by this action.
Browse: British Pharmacopoeia 2009 ДФУ
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Diazepam
Diazepam
General Notices
(Ph Eur monograph 0022)
C16H13ClN2Oээ284.7ээ439-14-5
DEFINITION
7-Chloro-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one.
Content
99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Very slightly soluble in water, soluble in ethanol (96 per cent).
IDENTIFICATION BP 2009
Infrared absorption spectrophotometry (2.2.24).
Comparison: diazepam CRS.
IDENTIFICATION BP 2007
A. Melting point (2.2.14):
B. Protect the solutions from bright light and measure the absorbances immediately. Dissolve 25 mg in a 5 g/l solution of sulphuric acid R in methanol R and dilute to 250.0 ml with the same acid solution (solution A). Dilute 5.0 ml of solution A to 100.0 ml with a 5 g/l solution of sulphuric acid R in methanol R. Examined between 230 nm and 330 nm (2.2.25), the solution shows 2 absorption maxima, at 242 nm and 285 nm. The specific absorbance at the maximum at 242 nm is about 1020. Dilute 25.0 ml of solution A to 100.0 ml with a 5 g/l solution of sulphuric acid R in methanol R. Examined between 325 nm and 400 nm, the solution shows a single absorption maximum at 366 nm. The specific absorbance at the maximum is 140 to 155.
C. Dissolve about 10 mg in 3 ml of sulphuric acid R. The solution shows greenish-yellow fluorescence in ultraviolet light at 365 nm.
D. To 80 mg in a porcelain crucible add
TESTS BP 2009
Related substances
Liquid chromatography (2.2.29). Prepare the solutions protected from bright light.
Test solution. Dissolve 25.0 mg of the substance to be examined in 0.5 ml of acetonitrile R
and dilute to 50.0 ml with the mobile phase.
Reference solution (a). Dilute 1.0 ml of the test solution to 100.0 ml with the mobile phase.
Dilute 1.0 ml of this solution to 10.0 ml with the mobile phase.
Reference solution (b). Dissolve the contents of a vial of diazepam for system suitability CRS (containing impurities A, B and E) in 1.0 ml of the mobile phase.
Column:
— size: l =
— stationary phase: spherical end-capped octylsilyl silica gel for chromatography R (5 µm);
— temperature:
Mobile phase. Mix 22 volumes of acetonitrile R, 34 volumes of methanol R and 44 volumes of a 3.4 g/l solution of potassium dihydrogen phosphate R previously adjusted to pH 5.0 with dilute sodium hydroxide solution R.
Flow rate. 1.0 ml/min.
Detection. Spectrophotometer at 254 nm.
Injection. 20 µl.
Run time. About 4 times the retention time of diazepam.
Identification of impuritiesэUse the chromatogram supplied with diazepam for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peaks due to impurities A, B and E.
Relative retention. With reference to diazepam (retention time = about 9 min): impurity E = about 0.7; impurity A = about 0.8; impurity B = about 1.3.
System suitability. Reference solution (b):
— resolution: minimum 2.5 between the peaks due to impurities E and A and minimum 6.0 between the peaks due to impurity A and diazepam.
Limits:
— correction factors: for the calculation of content, multiply the peak areas of the following
impurities by the corresponding correction factor: impurity B = 1.3; impurity E = 1.3;
— impurities A, B, E: for each impurity, not more than the area of the principal peak in the
chromatogram obtained with reference solution (a) (0.1 per cent);
— unspecified impurities: for each impurity, not more than the area of the principal peak in
the chromatogram obtained with reference solution (a) (0.10 per cent);
— total: not more than twice the area of the principal peak in the chromatogram obtained
with reference solution (a) (0.2 per cent);
— disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained
with reference solution (a) (0.05 per cent).
Heavy metals (2.4.8)
Maximum 20 ppm.
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on
Sulphated ash (2.4.14)
Maximum 0.1 per cent, determined on
ASSAY
(BP). Non-aqueous acidimetry, direct titration
Dissolve
1 ml of
STORAGE
Protected from light.
Action and use
Benzodiazepine.
Preparations
Diazepam Injection
Diazepam Oral Solution
Diazepam Rectal Solution
Diazepam Tablets
Ph Eur

Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Oxazepam
Oxazepam
General Notices
(Ph Eur monograph 0778)
C15H11ClN2O2
ээ286.7ээ604-75-1
DEFINITION
(3RS)-7-Chloro-3-hydroxy-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one.
Content
99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance
White or almost white, crystalline powder.
Solubility
Practically insoluble in water, slightly soluble in ethanol (96 per cent).
IDENTIFICATION
Infrared absorption spectrophotometry (2.2.24).
Comparisonэoxazepam CRS.
TESTS
Related substances
Liquid chromatography (2.2.29).
Prepare the solutions immediately before use.
Test solution Dissolve 40.0 mg of the substance to be examined in 25 ml of a mixture of equal volumes of acetonitrile R and water R and dilute to 50.0 ml with the same mixture of solvents.
Reference solution (a) Dilute 1.0 ml of the test solution to 100.0 ml with a mixture of equal volumes of acetonitrile R and water R. Dilute 2.0 ml of this solution to 10.0 ml with a mixture of equal volumes of acetonitrile R and water R.
Reference solution (b) Dissolve the contents of a vial of oxazepam for peak identification CRS(containing impurities A, B, C, D and E) in 1.0 ml of the test solution.
Column:
—size: l =
—stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 µm) resistant to bases up to pH 11.
Mobile phase:
—mobile phase A: dissolve
—mobile phase B: acetonitrile R;
Flow rate 1.0 ml/min.
Detection Spectrophotometer at 235 nm.
Injection 10 µl.
Identification of impurities Use the chromatogram obtained with reference solution (b) and the chromatogram supplied with oxazepam for peak identification CRS to identify the peaks due to impurities A, B, C, D and E.
Relative retention With reference to oxazepam (retention time = about 15 min): impurity E = about 0.7; impurity A = about 0.8; impurity B = about 1.2; impurity C = about 1.4; impurity D = about 2.0.
System suitability Reference solution (b):
—resolution: minimum 1.5 between the peaks due to impurities E and A.
Limits:
—correction factors: for the calculation of content, multiply the peak areas of the following impurities by the corresponding correction factor: impurity A = 4.0; impurity B = 1.1;
—impurities A, B, C, D, E: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (a) (0.2 per cent);
—unspecified impurities: for each impurity, not more than 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.10 per cent);
—total: not more than 5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (1.0 per cent);
—disregard limit: 0.25 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.05 per cent).
Loss on drying (2.2.32)
Maximum 0.5 per cent, determined on
Sulphated ash (2.4.14)
Maximum 0.1 per cent, determined on
IMPURITIES
Specified impurities A, B, C, D, E.
A. (5RS)-7-chloro-5-phenyl-4,5-dihydro-1H-1,4-benzodiazepine-2,3-dione,
B. (3RS)-7-chloro-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl acetate,
C. 6-chloro-4-phenylquinazoline-2-carbaldehyde,
D. (2-amino-5-chlorophenyl)phenylmethanone,
E. 7-chloro-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one 4-oxide.
Ph Eur
ASSAY
Dissolve
1 ml of
STORAGE
Protected from light.
Action and use
Benzodiazepine.(ВР 2009)
Anxiolytic. (ВР 2007)
Preparation
Oxazepam Tablets
Ph Eur
Browse: British Pharmacopoeia 2009
British Pharmacopoeia Volume I & II
Monographs: Medicinal and Pharmaceutical Substances
Nitrazepam
Nitrazepam
General Notices
(Ph Eur monograph 0415)
C15H11N3O3
ээ281.3ээ146-22-5
DEFINITION
Nitrazepam contains not less than 99.0 per cent and not more than the equivalent of 101.0 per cent of 7-nitro-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, calculated with reference to the dried substance.
CHARACTERS
A yellow, crystalline powder, practically insoluble in water, slightly soluble in alcohol.
IDENTIFICATION
First identificationэA, C.
Second identificationэA, B, D, E.
Э
A. Melting point (2.2.14):
эB. Protect the solutions from light and measure the absorbances immediately.
Dissolve 25.0 mg in a 5 g/l solution of sulphuric acid R in methanol R and dilute to 250.0 ml with the same solvent. Dilute 5.0 ml of the solution to 100.0 ml with a 5 g/l solution of sulphuric acid R in methanol R. Examined between 230 nm and 350 nm (2.2.25), the solution shows an absorption maximum at 280 nm. The specific absorbance at the maximum is 890 to 950.
Э
C. Examine by infrared absorption spectrophotometry (2.2.24), comparing with the spectrum obtained with nitrazepam CRS.
эD. Dissolve about 20 mg in a mixture of 5 ml of hydrochloric acid R and 10 ml of water R. Boil for 5 min, cool and add 2 ml of a 1 g/l solution of sodium nitrite R. Allow to stand for 1 min and add 1 ml of a 5 g/l solution of sulphamic acid R and mix. Allow to stand for 1 min and add 1 ml of a 1 g/l solution of naphthylethylenediamine dihydrochloride R. A red colour is produced.
эE. Dissolve about 10 mg in 1 ml of methanol R, warming if necessary, and add 0.05 ml of dilute sodium hydroxide solution R. An intense yellow colour is produced.
TESTS
Related substances
Carry out the test protected from light. Examine by thin-layer chromatography (2.2.27), using silica gel GF254 R as the coating substance.
Test solution Dissolve
Reference solution (a) Dissolve 10 mg of aminonitrobenzophenone R in acetone R and dilute to 100 ml with the same solvent. Dilute 10 ml of the solution to 50 ml with acetone R.
Reference solution (b) Dissolve 10 mg of nitrazepam impurity A CRS in acetone R and dilute to 100 ml with the same solvent. Dilute 10 ml of the solution to 50 ml with acetone R.
Reference solution (c) Dilute 1 ml of the test solution to 20 ml with acetone R. Dilute 1 ml of this solution to 50 ml with acetone R.
Apply separately to the plate 10 µl of each solution. Develop over a path of
Heavy metals (2.4.8)
Loss on drying (2.2.32)
Not more than 0.5 per cent, determined on
Sulphated ash (2.4.14)
Not more than 0.1 per cent, determined on
IMPURITIES
A. 3-amino-6-nitro-4-phenylquinolin-2(1H)-one,
B. (2-amino-5-nitrophenyl)phenylmethanone.
Ph Eur
ASSAY
Dissolve
1 ml of
STORAGE
Store protected from light.
Action and use
Benzodiazepine. (ВР 2009)
Hypnotic. (ВР 2007)
Preparations
Nitrazepam Oral Suspension
Nitrazepam Tablets
Ph Eur

