Usage of immunological tests in diagnosis of infectious diseases.
Reactions based on Agglutination and precipitation phenomenen
Lysis and complement fixation test.
Agglutinins and the Agglutination Reaction
Agglutinins are antibodies capable of clumping the corresponding microbes by producing visible conglomerates. The addition of the corresponding immune sera to a suspension of microbes provokes clumping of microbial cells in the form of flakes or granules. This phenomenon is known as agglutination. The agglutination reaction takes place on mixing erythrocytes, yeasts and other cells with the corresponding immune sera.
It was described by A. Charrin, G. Roger (1889), V. Isaiev and V. Ivanov (1894) and was investigated in detail by M. Gruber and H. Durham in
typhoid bacteria. Later it was established that in a whole series of infectious diseases antibodies (agglutinins) are produced in the blood of patients, which are capable of clumping the corresponding causative agents of infectious diseases.

The agglutination reaction, like the flocculation and precipitin reactions, is under the control of the physicochemical conformities of the interrelations of colloidal systems. The antibody (agglutinin) and antigen (agglutinogen) take part in the agglutination reaction. Their interaction takes place in definite quantitative proportions, and in the presence of an electrolyte (0.85 per cent NaCI solution). In mechanism and outer manifestation the agglutination reaction is similar to the precipitin reaction. Both reactions are accompanied by the production of visible precipitates of antigen with the difference that in the agglutination reaction microbial bodies serve as the antigen, in the precipitin reaction the antigen is the product of the breakdown of microbial bodies, very minute particles of dissolved antigens requiring a large amount of antibodies for complete interaction.
The agglutination reaction is characterized by specificity, but group agglutination can be found, that is, the clumping of closely related microbes though in weaker serum dilutions.
The antigenic structure of bacteria is quite varied. In one and the same bacterial strain there may be group, species, and type antigens. Similar bacteria are composed of various antigenic groups, and during immunization of animals the corresponding agglutinins are produced in the blood. This can be represented in the table shown on the next page.
As may be seen from this table, the serum received against microbe A agglutinates microbe A readily, since agglutinins a1 b1 c1 completely correspond to the agglutinogens a b c. This serum agglutinates microbe B (to a lesser degree) due to the homologous b1 c1-agglutinins and bc-agglutinogens, and also microbe C (to an even lesser extent) due to the common character of c1-agglutinin and c-agglutinogen. These interrelations are found between the serum against microbe B and microbes B, C and A, etc.
The variety of antigens in microbial cells is a regular process and reflects the law of homologous series of in intraspecies and interspecies variability of bacteria (see section on variability of bacteria).
|
Species of bacteria |
A |
B |
C |
|
Antigens (agglutinogens) |
abc |
bed |
cde |
|
Antibodies (agglutinins) |
a1 b1 c1 |
b1 c1 d1 |
c1 d1 e1 |
Thus, upon immunization of the animal with one species of microbe agglutinins may occur not only to this species, but to other related bacterial species which have general group antigens.
For revealing specific agglutinins in sera of animals immunized by a complex of antigens of the bacterial cell the method of adsorption of agglutinins is employed (Castellani’s exhaustion reaction). By adding certain species of bacteria to the serum of an immunized animal, in which there are several agglutinins, those which clump only organisms of this species are removed, after which the serum freed from these agglutinins is checked for the presence of other agglutinins by adding other species of bacteria.
The method of agglutinin adsorption is used to study the antigenic structure of bacteria which are used for preparing agglutinating and therapeutic sera, vaccines, and diagnostic preparations. Agglutinating sera obtained by this method are called monoceptor sera. They make it possible to determine more precisely the species and type specificity of the causative agents of salmonellosis and dysentery. In motile microbes there are flagellar (H) and somatic (0) antigens. During immunization of animals with motile bacteria H-agglutinins and 0-agglutinins are correspondingly produced. Flagellar agglutinins cause a more rapid clumping of microbes in the form of loose flakes, while somatic agglutinins produce comparatively slowly conglomerates of bacteria in the form of fine granules. H-agglutination is otherwise known as large flaky and 0-agglutination as fine granular agglutination.
Bacteria containing the Vi-antigen are only weakly or eveot agglutinated by 0-sera, but agglutinate well with Vi-sera. This shows that 0- and Vi-antigens as well as 0- and Vi-antibodies have a different structure.
Slide agglutination test
The reaction of agglutination may take place as a result of the action of non-specific factors (without the presence of an agglutinating serum), the main colloidal solutions of dyes and acids. Such non-specific reactions may also take place in the presence of an isotonic solution alone m microbes which were exposed to considerable changes as a result of long storage, and also in R-forms of bacteria. The extent of manifestation of the specific agglutination reaction depends on the salt concentration (electrolyte), serum concentration, density of bacterial suspension, pH, influence of temperature, shaking and mixing, etc.
The agglutination reaction is widely employed in the practice of serological diagnosis of enteric fever, paratyphoids A and B {Widal’s reaction), brucellosis (Wright’s reaction), typhus fever reaction with Rickettsia prowazeki), tularaemia, leptospirosis and other diseases, in which with the help of known microbes (diagnosticums) the corresponding agglutinins are determined in patients’ sera. For determining Vi-antibodies in carriers of enteric fever salmonellae Vi-agglutination has had wide application in laboratory diagnosis. The agglutination reaction is used for the identification of isolated microbes in patients and sick animals with the application of previously known agglutinating sera.
To obtain a quick response accelerated agglutination reactions are used as tentative methods in some cases Nobel’s reaction for detecting typhus and enteric fever, Huddleson’s reaction for brucellosis, Minkevich’s reaction for typhus fever and tularaemia and the agglutination reaction with luminescent sera for revealing causative agents of intestinal infections, anthrax, etc.
In surgical practice of blood transfusion the reaction of isohaemagglutination has had wide application with the help of which blood groups may be determined. For this purpose it is necessary to have two haemolytic sera (β and γ) obtained from people with A and B blood groups. One or two drops of each of these sera are put separately on a slide or china dish, and one small drop of the blood under test is added. The blood and serum are carefully mixed and, according to the reaction of isohaemagglutination, the blood group is established.
The agglutination reaction may also take place without the participation of antibodies. Some plants (leguminous) were found to contain haemagglutinins which agglutinate human erythrocytes of definite blood groups; phytohaemagglutinins have been revealed in saline solutions of the fruits and seeds of definite types of plants; they are used in haematological studies.
To obtain agglutinating animal sera (rabbit, etc.), the animals are immunized with a suspension of freshly isolated bacteria of a certain species or type according to a certain schedule, taking into account the dose and the intervals between vaccinations. At the end of immunization blood is taken from the animals and the serum obtained is inactivated, conserved and titrated. The titre of the agglutinating serum is known as the smallest amount or the greatest dilution which causes a clearly marked agglutination reaction. On the labels of ampoules of manufactured sera the titres are written as fractions indicating the maximum dilution (1 : 3200, 1 : 6400, 1 : 12800, 1 : 25600, etc.) at which they cause agglutination of the corresponding antigen (agglutinogen). Agglutinating sera are produced as non-adsorbing and adsorbing, multivalent, species and type specific.
Serological examination.
All immunological tests are based on specific antibody-antigen interaction. These tests are called serological since to make them one should use antibody-containing sera.
Serological tests are employed in the following cases: (a) to determine an unknown antigen (bacterium, virus, toxin) with the help of a known antibody; (b) to identify an unknown antibody (in blood serum) with the help of a known antigen. Hence, one component (ingredient) in serological tests should always be a known entity.
The main serological tests include tests of agglutination, precipitation, lysis, neutralization, and their various modifications.
Agglutination Test. The term agglutination means clumping of microorganisms upon their exposure to specific antibodies in the presence of electrolyte. The presumptive and standard agglutination tests (AT) are widely utilized in the diagnosis of numerous infectious diseases.
To perform agglutination tests, one needs three components: (1) antigen (agglutinogen); (2) antibody (agglutinin); (3) electrolyte (isotonic sodium chloride solution).
Standard agglutination test is employed for determining the serogroup and serovar of microorganisms and is performed according to the scheme presented in Table 1. All ingredients are dispersed into test tubes in a definite sequence. Serum is diluted in simple numerical ratios such as 1:100, 1:200, 1:400, etc.
Into each tube with diluted serum, transfer 1-2 drops of the antigen (1-2 milliard microorganisms per ml), shake vigorously, and place into a
A negative test (–), there is no sediment, the suspension remains uniformly turbid, showing no difference from the content of the test tube with the antigen control.
External manifestations of the agglutination test depend on the type of antigen and the size of cells. In bacteria, the interaction between somatic antigens (0-antigens) and specific antibodies is slow and a fine granular sediment forms in 18-20 hrs. Small grains of the agglutinate do pot break upon shaking. Such agglutination is observed in bacteria of tularaemia, Brucella, etc. The presence of the flagellar H-antigen (Salmonella of typhoid, paratyphoid, food tox-infections) induces a rapid development of agglutination. Readily breakable large loose flocculi form in 2-4 hrs (Fig. 1).
Table 1
Schematic Representation of the Agglutination Reaction
|
Ingredient |
Number of the test tube |
||||||
|
1 |
2 |
3 |
4 |
5 |
6 antigen control |
7 serum control |
|
|
Isotonic sodium chloride solution, ml |
1 |
1 |
1 |
1 |
1 |
1 |
– |
|
The patient’s serum in a 1: 50 dilution, ml |
1® |
1® |
1® |
1® |
1 |
– |
1 |
|
The obtained dilution of the serum |
1:100 |
1:200 |
1:400 |
1:800 |
1:1600 |
– |
1:50 |
|
Bacterial suspension, drops |
2 |
2 |
2 |
2 |
2 |
2 |
– |
|
Incubation at 37 |
|||||||
Figure 1. Agglutination test
Agglutination of living Leptospira is studied in wet-mount preparations with lateral illumination. Agglutinated Leptospira appear as luminescent “spiders” against a dark background.
The use of the AT in the serological diagnosis of infectious diseases, such as typhoid and paratyphoids (Widal’s reaction), epidemic typhus (Weigl’s reaction), brucellosis (Wright’s and Huddleson’s reaction), tularaemia and other diseases, is based on determining antibodies (agglutinins) in the patient’s serum.
To perform the test, take 3-5 ml of blood from the jugular vein, finger or earlobe in au adult or 1 ml from the heel in small children. Separate the serum from blood and dilute it. with isotonic saline in a ratio blood may contaiormal antibodies which are capable of inducing agglutination reaction in small dilutions.
As to the antigen that is utilized in this test, diagnosticums (suspensions of known killed and occasionally living microorganisms) are employed for this purpose. Diagnosticums of killed microorganisms are fairly stable, retaining their properties for several years and present no risk of contamination.
The procedure of the AT with the patient’s blood and evaluation of the results do not differ from those used in the standard AT aimed at determining the species of microorganisms.
Indirect agglutination (haemagglutination) (IHA) test. Occasionally, antigens employed for the agglutination reaction are so highly dispersed that an agglutinogen-agglutinin complex evades detection by the naked eye. To make this reaction readily visible, methods of adsorption of such antigens on larger particles with their subsequent agglutination by specific antibodies have been designed. Adsorbents employed for this purpose include various bacteria, particles of talc, dermal, collodium, kaolin, carmine, latex, etc. This reaction has beeamed indirect (or passive) agglutination test.
Red blood cells display the highest adsorptive capacity. The test conducted with the help of erythrocytes is called indirect, or passive, haemagglutination (IHA or PHA). Sheep, horse, rabbit, chicken, mouse, human, and other red blood cells can be used for this test. These are prepared in advance by treating them with formalin or glutaraldehyde. The adsorptive capacity of erythrocytes augments following their treatment with tannic or chromium chloride solutions.
Antigens usually used in the IHA test are polysaccharide antigens of microorganisms, extracts of bacterial vaccines, antigens of viruses and Rickettsia, as well as other protein substances.
Erythrocytes sensitized with antigens are called erythrocytic diagnosticums. Most commonly used in preparing erythrocytic diagnosticums are sheep red blood cells possessing high adsorptive activity.
Procedure. Blood taken from the jugular vein of an adult sheep is placed into a glass jar with beads, defibrinated by shaking for 10-15 min, and filtered through a cotton-gauze filter. Following 10-minute centrifugation at 2000 X g, red blood cells are washed 3-4 times in isotonic saline solution, and the sediment is resuspended in the same solution. Then, a five-fold volume of 4 per cent formalin (pH 7.0) is added to the jar and erythrocytes are left to stand at 4 °C for 3-4 days. The erythrocytes are precipitated once again and the procedure is repeated with a fresh solution of formalin. After this the red blood cells are washed with a 20-fold volume of physiological saline and adjusted to 20 per cent concentration. The fixed red blood cells are kept at
The suitability of erythrocytes is checked by the following criteria: (1) no haemolysis should be observed after freezing and thawing of 5 per cent erythrocyte suspension in distilled water; (2) mixing of 0.1 ml of 0.2 per cent suspension of erythrocytes with isotonic NaCI solution brings about no spontaneous agglutination.
To sensitize red blood cells, to eight volumes of distilled water add one volume of antigen, one volume of 50 per cent suspension of formalin-treated erythrocytes, and one volume of 0.1-0.2 per cent solution of chromium chloride and tannin in dilution 1:20000-1:2000000.
Allow the mixture to stand for 10-15 min at room temperature; then add an equal amount of isotonic saline and centrifuge it for 20 min at 2000 X g. The sediment of sensitized erythrocytes is washed two-three times with a 20-fold volume of physiological saline solution, then resuspended to 5 per cent concentration in the stabilizing solution consisting of equal volumes of 30 per cent solution of sucrose and human donor blood.
As a control, use formalin-treated red blood cells sensitized by another antigen or formalin-treated non-sensitized red blood cells.
It is convenient to set up an indirect haemagglutination test on micropanels of the Takata apparatus using a microtitrator for diluting the material. The sera to be assayed are heated for 30 min at
The results of the test are assessed by the presence of haemagglutination. It is considered positive if the titre of haemagglutination with the erythrocytes assayed exceeds by at least four times the titre of haemagglutination with the control erythrocytes. The sensitized red blood cells should be invariably checked for the absence of spontaneous agglutination.
|
I. Components: 1. Pair serum from patient (unknown Ab)
2.Specific erythrocytes diagnosticum
3.NaCl solution |
|
|
|
|
||
|
Delution of serum from patient Розведення сироватки хворого 1:20 1:40 1:80 1:160 1:320 Контроль Control
Agglutination Аглютинація Non agglutination Немає аглютинації |
||
|
|
||
|
II. Реакція гальмування гемаглютинації Hemagglutination inhibition test
|
||
Reaction with incomplete antibodies (Coombs test).
Interrelations between protective (defensive) antigens and the corresponding antibodies are of a completely different nature. During this interactioo typical immunological reactions are observed (neutralization, precipitation and complement–fixation). It has been suggested that protective antigens provoke the formation of incomplete or blocking antibodies capable of rendering harmless the aggressins of anthrax bacilli, capsular proteins of the causative agents of plague, tularaemia and of other bacteria.
Incomplete (monovalent) or blocking antibodies are fixed by the antigens, but do not cause their agglomeration. In contrast to ordinary (complete) antibodies they proved to be more stable to heat, pressure, and chemicals, and quite easily penetrate through the placenta. They include rhesus-agglutinins, non-precipitating thermolabile antibodies and reagins of allergic patients, and of patients with systemic lupus, infectious polyarthritis, and collagenosis. Incomplete hetero-, iso-, and autoantibodies may cause drug leuko- and thrombocytopenia.
Incomplete agglutinins and haemagglutinins have been demonstrated in immunization of animals with the capsular antigen of the causative agent of plague. They were found in the sera of patients suffering from dysentery, typhus, and brucellosis in titres 4-32 times those of complete antibodies; in the sera of animals immunized with the cholera vaccine their titre was 4-8 times that of complete antibodies.
Of most interest are agglutinins against the rhesus-antigens of erythrocytes of children suffering from haemolytic disease which is the result of the presence of a rhesus-factor in the erythrocytes inherited from the father. After penetrating into the blood of the mother the rhesus-factor provokes the production of rhesus-agglutinins which later enter the blood of the foetus through the placenta and cause agglutination of erythrocytes. Haemolytic disease is due to the incompatibility of the rhesus-factor in the blood of the mother and the foetus.
The rhesus-factor is capable of causing the production of two types of agglutinins: (1) complete (bivalent) agglutinins which in a saline and colloidal medium may cause the agglutination reaction of erythrocytes containing a particular antigen, and (2) incomplete (monovalent) agglutinins inhibiting agglutination, which do not cause the agglutination reaction in a saline solution.
For detecting incomplete antibodies special methods are employed. The Coombs’ test is used, in particular to detect incomplete agglutinins in rhesus-negative mothers. To determine the fixation of agglutinins by the patients’ erythrocytes, antiglobulin serum is added, which, in a saline solution, is capable of causing marked agglutination of erythrocytes sensitized by incomplete agglutinins. A molecule of antiglobulin binds two molecules of incomplete agglutinins fixed to two different erythrocytes, due to which the agglutination reaction takes place.
Coombs’ test represented schematically (1–direct; 2– indirect)
The direct reaction (Fig. 1, left) demonstrates the presence in the patient’s blood of antibodies bound with the erythrocytes by means of antiglobulin: in the indirect reaction (Fig. 1, right) free incomplete antibodies are revealed by adding to the serum normal erythrocytes, bacteria or rickettsia and then antigamma globulin.
|
|
|
Reversed indirect haemagglutination (RIHA) test is used for detecting bacterial and viral antigens in the materials to be examined as well as for the rapid diagnosis of a number of infections.
In contrast to IHA. erythrocytes in this test are sensitized not by antigens but by antibodies whose agglutination occurs upon addition of the antigen.
Erythrocytes are first fixed with formalin or glutaraldehyde and bound to gamma-globulin which is isolated from immune sera and purified from other serum proteins. Binding of gamma-globulin with the erythrocyte surface is mediated by chromium chloride. For this purpose, to 8 volumes of distilled water add 1 volume of immunoglobulins obtained from immune serum, 1 volume of 50 per cent suspension of formalin-treated red blood .cells, and 1 volume of 0.1-0.2 per cent solution of chromium chloride. Allow the mixture to stand at room temperature for 10-15 min, thetreat the erythrocytes as in the passive haemagglutination test.
This test is commonly used to identify the causative agents in post-mortem material taken from the organs of man and animals, for example, from the brain, spleen, liver, and lungs. Prepare 10 per cent suspension of the above organs with isotonic sodium chloride solution, centrifuge it at 10 000 X g for 30-60 min, and use the supernatant as an antigen.
Procedure. Prepare two-fold dilutions of the material to be studied (antigen) with a stabilizing solution. Place one drop of each dilution of the antigen into 3 neighbouring wells of the micropanel (the reaction requires 3 parallel rows of wells). Into each well of the first row add 1 drop of the stabilizing solution, into wells of the second row, 1 drop of homologous immune serum in a 1:10 dilution, those of the third row, 1 drop of heterologous immune serum. The second and third rows serve as controls of reaction specificity. Let the mixture stand at room temperature for 20 min.
To all wells add one drop of 1 per cent suspension of sensitized erythrocytes (erythrocyte antibody diagnosticum) and shake the plates well. Read the results of the reaction in 30-40 min. In the presence of the specific antigen, haemagglutination is observed in the first and third rows (with heterologous serum) and is absent in the second row -where the antigen is preliminarily neutralized by homologous serum.
To ensure the accuracy of the test, the sensitized red blood cells are checked for spontaneous agglutination.
Reversed indirect haemagglutination inhibition (RIHAI) test makes it possible to detect the presence of antibodies in human and animal sera.
Procedure. Dilute sera by ten-fold with isotonic sodium chloride solution, heat for 20 min at
The test is validated by checking whether sensitized erythrocytes may undergo spontaneous agglutination in the presence of: (a) stabilizing solution; (b) normal antigen (from material free of the virus); (c) the serum tested. Among advantages of the test one can cite its universality and the possibility to use it for finding various antigens.
Assessment of the haemagglutination results. Estimation of the results of IHA, BIHA and RIHAI tests is relied on the degree of erythrocyte agglutination; (++++)> complete agglutination; (+++), almost complete agglutination; (++), partial agglutination; (+), traces of agglutination; (–), no agglutination.
The test is considered positive, if agglutination is complete (++++) or almost complete (+++)i the diagnosticum does not induce spontaneous agglutination in the presence of each component required for the reaction, and the control test of the specificity of the antigen or antibody is positive.
Recent years have seen wide application of serological reactions which make use of antibodies or antigens labelled in some way. The label may be different but should meet the basic requirement: it should be easily detected by means of definite reactions or under the microscope. Apart from retaining the specificity of immunological reactions, serological reactions with labels make it possible to rapidly obtain the results and are usually distinguished by high sensitivity. It is not unnatural, therefore, that they have found widespread application for the rapid diagnosis of viral and bacterial infections.
Latex agglutination test: latex beads coated with specific antibody are agglutinated in the presence of the homologous bacteria or antigen. This test is used to determine the presence of the H. influenzae, N meningitidis, several species of streptococci, and the yeast Cryptococcus neoformans.
Latex agglutination test
Immunofluorcscence (or Coons’ reaction).
The following types of labels are employed most frequently: (1) fluorochromes capable to fluoresce in ultraviolet rays or the blue-violet area of the spectrum of visible light, for example, fluoresce-inisothiocyanate (FITC) which is utilized for performing immunofluorescent reactions; (2) ferritin, a protein containing up to 23 per cent of iron, which is readily visible by electron microscopy and thus is well suitable as a label in immunoelectron microscopy; (3) enzymes which induce the breakdown of the substrate with the formation of stained products when they get in contact with the substrate; they are used for performing immunoenzymic reactions; (4) radioactive labels utilized in highly sensitive radioimmunoassays.
The above serological tests vary in their sensitivity and diagnostic value. Some of them (e.g., radioimmunoassay) require the employment of sophisticated recording equipment, as well as special measures of protection from radiation.
Immunofluorescence (IF) test relies on the utilization of FITC or other fluorochromes which are chemically conjugated with antibodies. The antibodies labelled by FITC (as part of immunofluorescent sera) retain the immunological specificity and interact with strictly delinite antigens. Antibody-labelled antigen complexes are readily recognized by intense yellow-green fluorescence during examination with the luminescent microscope.
There are several variants of immunofluorcscence (or Coons’ reaction).
1. Direct immunofluorescence envisages the employment of immunofluorescent sera against each antigen tested.
2. Indirect immunofluorescence is based on the use of two sera. First, unlabelled antibodies against the antigen to be assayed are utilized. At the second stage, the formed antigen-antibody complex is treated with FITC-labelled serum containing antibodies against gamma-globulins of that species of animals from whom the antisera used at the first stage of the reaction were obtained (anti-species serum).
For example, if the serum employed at the first stage of the reaction has been obtained by rabbit immunization, at the second stage one employs labelled anti-species rabbit serum obtained by immunizing with rabbit gamma-globulins of donkeys or some other animals. In this case antiglobulin-labelled antibodies coat the antigen tested with a second layer (the first one has been formed by unlabelled antibodies which, in turn, serve as antigens for antiglobulin (anti-species) serum). As a result, the antigen becomes visible under the luminescent microscope.
Immunofluorescence may be used for studying various antigens: cultures of bacteria, fungi, Protozoa, preparations from patients’ material; infected cultures of cells, tissue sections, etc. The material to be examined is placed on a glass slide and fixed (most often in acetone for 10 min at room temperature) after which it is dried for 20 min at
Figure. 2 Fluorescein labeled antibody test methods Note that on must have fluorcscein-labeled antibody that is specific rur the organist in question in order to use the direct method, however, the indirect method can be adapted to any organism using fluorcscein labeled ant human gamma globulin as the only labeled antiserum.
In direct immunofluorescence the preparation is stained with specific labelled serum in a humid chamber for 30 min at
In indirect immunofluorescence the preparation is first treated with unlabelled specific serum in a humid chamber for 30 min at
Ready preparations are dried with filter paper and examined under a luminescent microscope, first, with a dry objective (40 x), then, after having placed on the preparation a drop of non-fluorescent oil, with the immersion objective (90 x). Attention is paid not only to the presence of green or green-yellow luminescence, but also to its intensity and the arrangement of luminescence in the cell examined.
To rule out false positive results, a number of control techniques is available. Of particular significance among them is a control with a heterologous antigen (for example, with bacterial culture which does not correspond to the serum tested from an antigcnic standpoint). In studying infected cultures of cells, a control with normal noninfected culture should be used (to exclude autofluorescence and non-specific binding of labelled antibodies with the cell surface). To inhibit auto fluorescence of the preparation, one can use bovine albumin labelled with sulpharodamine.
While retaining the pecificity of immunological tests, the mmunofluorescence reaction is distinguished by its simplicity and rapidity. Indirect immunofluorescence may be employed not only for investigating antigens but also for determining the number of antibodies in immune serum. On the other hand, the IF test cannot be considered as a highly sensitive reaction. Furthermore, non-specific adsorption of labelled antibodies on the preparation with the appearance of false positive results is also possible.
Fluorescein-labeles antibody. Fluorescein-labeled antibodies are used in a procedure for rapidly determining the presence of specific antigens or antibodies to a known antigen. As illustrated in Figure2, the indirect method can be used to detect the presence of antibodies to any bacterium. All that is required is known bacteria, the patient’s serum, and some fluorescein-labeled antihuman y-globulin.
The direct method, also illustrated in Figure 2, can be used to confirm a tentative identification of an isolated organism. In this case, however, the microbiologist is limited by the availability of fluorescein-labeled specific antibody to the bacterium in question. The commercial availability of many different monoclonal antibodies has facilitated the rapid identification of many bacteria and viruses. Such antibodies can be selected to provide a much higher specificity than can be obtained with a heterologous population of antibodies. Monoclonal antibodies can be linked to fluorescent dyes and used to detect microorganisms in tissues and infected cell cultures.
Direct method: 1 Antigen. 2.Cover with fluorescein-labeled group A streptococcus antiserum and incubate 3.Wash away the excess fluoroscein-labeled antiserum and examine.
Indirect method: 1.Antigen. 2.Cover with unlebeled serum from patient with active syphilis and incubate 3. Wash away the excess unlabeled patient’s antiserum. 4. Cover with fluoroscein-labeled anti-human gammaglobulin antiserum and incubate. 5.Wash away the excess fluoroscein-labeled antiserum and examine.
Precipitins and the Precipitin Reaction. Precipitins are antibodies which bring about the formation of a minute deposit (precipitate) upon interaction with a specific antigen. The precipitin reaction is a specific interaction of the antigen (precipitinogen) and antibody (precipitin) in the presence of an electrolyte (0.85 per cent NaCI solution) with the formation of a deposit or precipitate. In 1897 R. Kraus roved that the transparent filtrate of a broth culture of plague bacteria became turbid when mixed with an antiplague serum with the subsequent falling-out of flakes to the bottom of the test tube. An analogous phenomenon was observed upon the interaction of cholera and typhus culture filtrates with the corresponding sera. R. Kraus named this a precipitin reaction, and the antibody producing it – precipitin. After the injection of eel serum protein into rabbits in
In mechanism this reaction is similar to the flocculation reaction and the physicochemical interrelations of highly dispersed colloids form the basis for it.
The precipitin reaction is specific and sensitive. It allows the detection of antigen precipitinogen) in a dilution up to 1 : 1000000 and 1 : 10000000.
Precipitinogens during parenteral injection provoke the formation of specific precipitins in the body and combine with them. Proteins of animal, plant and microbe origin: blood, serum, extracts from different organs and tissues, foodstuffs of a proteiature (meat, fish, milk), filtrates of microbial cultures or affected tissues, may all be used as precipitinogens.
Precipitinogens of the causative agents of anthrax, plague and tularaemia are thermoresistant. Some precipitinogens withstand heat up to 120-180 °С.
The precipitin reaction is used in the diagnosis of anthrax, tularaemia, etc., in the typing and studying of antigen structure of certain groups of bacteria. In forensic medicine with the aid of the precipitin reaction the origin of blood spots and sperm is determined, in sanitary examination an admixture of milk of one species of animal to another is revealed, the addition of artificial honey to natural, the falsification of meat, fish and flour goods, etc., and in biology the genetic links between related species of animals, plants and micro-organisms are established.
The precipitin reaction is most widespread in the diagnosis of anthrax (Ascoli’s test) for detecting the antigen of anthrax bacilli in extracts from the organs of animals, skin, wool, hair and also for the control of the manufactured goods: fur jackets, fur collars, shaving brushes and other goods. This reaction is known as the thermoprecipitin reaction, since the extract which undergoes investigation is preliminarily boiled, then filtered to obtain a transparent solution and layered on the precipitating anthrax serum. The thermoprecipitin reaction {ring precipitation} is widely used for the diagnosis of plague and tularaemia during the investigation of extracts prepared from the internal organs (spleen, liver) of the cadavers of wild rodents.
In special laboratories there is a set of precipitating species-specific sera which are obtained by long-term immunization of animals with the corresponding antigens (precipitinogens). At the end of the immunization course blood is taken from animals (rabbits, donkeys, sheep, goats), a serum is obtained and the strength of its action is determined
The titre of the precipitating serum is known as the maximum dilution of antigen (precipitinogen) in which a clearly expressed precipitin reaction is obtained. Whole precipitating serum is taken because the dispersion of the precipitating serum is less in comparison to that of the precipitinogen. For this reason in order to obtain optimal quantitative proportions of particles of acting components the antigen and not the serum is diluted.
In order to differentiate antibodies against different antigens, the method of the diffusion precipitin reaction with antiserum mixed with gelatin or agar is used. After the antigen solutions are layered easily discernible precipitation zones occur within this agar, each specific pair of antigen-antibody complex having its own zone. This method was later improved. In wells made in agar in Petri dishes solutions of antigens and antibodies are poured which diffuse into the gel, and after coming in contact with one another form lines of precipitation. The merging of the ends of precipitating lines provides evidence for the similarity of antigens of comparative systems. At present there are several modifications.
The precipitin reaction can be carried out on paper. If the mixture of antigens is isolated by the method of paper electrophoresis and then the strips of paper are treated with immune serum, then a precipitate is produced in definite places, the localization of which determines the nature of antigen if the origin of the antibody is known, and vice versa. The precipitin reaction became the basis of the method of immunoelectrophoresis proposed by P. Grabar and K. Williams. At first, the antigen is separated in the electric field, after which it is developed by antiserum poured in a groove running parallel to the line along which the antigens moved during electrophoresis. Each antigen gives an individual band with the antibody. From the number and arrangement of the bands one can determine the presence of these or other antigens in the solution under test. With the help of immunoelectrophoresis antigen fractions which were unknown earlier have been revealed in various complexes. It allows to detect pathological deviations in the sera of patients. The immunoradiographic method is very effective.
Modifications of the Precipitin Reaction. Several modifications of the classic precipitin reaction have been developed. Such changes are designed either to increase the sensitivity of the reaction or to identify specific Ag-Ab reactions occurring in a system containing multiple Ags and Abs.
Double diffusion (Ouchterlony technique). When soluble Ag and soluble Ab are placed in separate small wells punched into agar that has solidified on a slide or glass plate, the Ag and the Ab will diffuse through the agar. The holes are located only a few millimeters apart, and Ab and Ag will interact to form a line of precipitate in the area in which they are in optimal proportions. Because different Ags diffuse at different rates, and because different Ags can require different concentrations of Ab for optimal precipitation, the position of the precipitin band usually varies for each Ag. In specimens containing several soluble Ags, multiple precipitin lines are observed, each occurring between the wells at a position that depends on the concentration of that particular Ag and its Ab (Fig.1). Thus, this simple diffusion method can be used to detect the presence of one or more Ags or Abs in a clinical specimen. In addition, if set up differently, this gel method can be used to detect similarities or differences in Ags. For example, Figure 2 shows the use of various Abs to distinguish between human IgM and IgG, and to detect specific epitopes on human IgG
Radial immunodiffusion. Radial immunodiffusion is used to measure the amount of a specific Ag present in a sample and can be used for many Ags. The most widely used diagnostic application of this procedure is to measure the amount of a specific Ig class present in a patient’s serum. The assay is carried out. by incorporating monospecific antiserum (antiserum containing only Ab to the Ag being assayed for) into melted agar and allowing the agar to solidify on a glass plate in a thin layer. Holes then are punched into the agar, and different dilutions of the Ag are placed into the various holes. As the Ag diffuses from the hole, a ring of precipitate will form at that position where Ag and Ab are in optimal proportions (Fig. 3). The more concentrated the Ag solution, the farther it must diffuse to be in optimal proportion with the constant Ab concentration in the agar gel. Thus, the diameter of the precipitin ring is a quantitative measure of Ag concentration. Using known concentrations of the Ag in question, a standard curve can be prepared by plotting the diameter of the precipitin ring versus Ag concentration.
Once a standard plot is obtained, the diameter of the precipitin ring formed with the unknown Ag can be measured to calculate its concentration.
Nephelometry is supplanting radial immunodiffusion as a method of measuring various Ig classes present in a patient’s serum. As discussed earlier, when Abs to various classes of Igs are mixed with serum, Ag-Ab complexes form and create a precipitate in a previously clear solution. Tubes containing known concentrations of Abs to Igs are incubated with different volumes of a patient’s serum. The precipitation reaction, seen as a cloudiness, is measured in an instrument called a nephelometer. This instrument interprets the Ag-Ab precipitates as increased light scattering compared to a control tube containing no precipitate. As with the radial immunodiffusion procedure, iephelometry, standard curves are performed using Ig class standards of known concentrations and Abs to the various Ig classes.
Through a certain concentration range, it is possible to generate a straight line plot when Ig concentration is plotted as a function of the amount of light scattering indicated by the nephelometer. This technique is being used increasingly in hospital laboratories and clinics as a quantitative measure of serum Ig class concentration in patient blood.
FIGURE 3 Radial immuno-diffusion This test is based on a precipitate that is formed as antigen diffuses into a semisolid medium that contains antibody Because the amount of antibody in the agar bed is constant, the diameter of the precipitin ring formed is a function of the amount of antigen applied A. Ann IgA is incorporated into the agar and small wells are punched out, into which patient serum or a standard antigen solution is placed The top row consists of standard amounts of known IgA The wells in the bottom row receive sera from four different patients B. A curve is drawn by plotting the diameter of the precipitin rings formed by the standard IgA solutions against the logarithm of the concentration of the standard IgA The concentration of IgA in each patient’s serum then is read from this standard curve after measuring the diameter of the ring formed by the patient’s serum in the bottom row.
Precipitation Test. The term precipitation test (PT) refers to sedimentation from the solution of the antigen (precipitinogen) upon its exposure to immune serum (precipitin) and electrolyte. Using the precipitation testю Using the precipitation test (PT), one can demonstrate the antigen in such tiny amounts which cannot be detected by chemical techniques.
Conduction of the PT requires liquid and transparent antigens representing ultramicroscopic particles of colloid solution of protein, polysaccharides, etc. Antigens are represented by extracts from microorganisms, organs, and tissues, products of breaking down of microorganism cells (lysates, filtrates). Resistance of precipitinogens toward a high temperature is used for for obtaining antigens from the causative agents of anthrax, plague (the boiling technique).Precipitating sera are prepared in a batch manner by hyperimmunization of animals (rabbits) with bacterial suspension, filtrates of broth cultures, autolysates, salt extracts of microorganisms, serum proteins, etc.
The titre of the precipitating serum, in contrast to the titre of other diagnostic sera, is determined by the maximum dilution of the antigen which is precipitated by a given serum. This is explained by the fact that the antigen participating in the precipitation reaction has an infinitesimal magnitude and that in a volumetric unit of the serum there are much more antigens than antibodies. Commercially available precipitating sera have the titre of no less than 1:100 000.
Procedure. In a narrow test tube (
Precipitation test: 1 – in a test tube; 2—in gel; 3 –immunoelectrophoresis (order of picture: left–right–down )
The precipitation test is widely employed in the laboratory practice for the diagnosis of infectious diseases of the bacterial (anthrax, plague, tularaemia) and viral (natural small pox, acute respiratory infection) nature.
The results of the test are read in 5-10 min, 1-2 hrs or 20-24 hrs, depending on the type of an antigen and antibodies. If the reaction is positive, a precipitate in the form of a white ring forms on the borderline between the serum and the extract tested.
In forensic medicine the precipitation test is employed for classifying the species of the protein (blood stains, sperm).
The PT may demonstrate not only species but also group specificity of the protein. Thus, the degree of homogeneity of various species of animals and plants has been determined with its help.
The use of the precipitation test for sanitary and hygienic control of foods luns makes it possible to uncover adulteration of meat, fish, and flour products, as well as admixtures in milk. etc.
Disadvantages of the PT are instability of the precipitate (the ring) which disappears even upon the slightest shaking and impossibility to establish the number of various antigens participating in the formation of the precipitate.
The precipitation reaction in gel is free of these disadvantages. Test of precipitation in gel (PG) is based on the interaction of homologous antibodies and antigens in an agar gel and the formation of visible bands of precipitation. As a result of counter-diffusion into gel, the antibodies and antigen form immune complexes (aggregates) visualized in the form of opalescent (white) bands (Fig.4).
When several antigens diffusing irrespective of each other are present, the number of bands corresponds to the number of antigens. Serologically homogeneous antigens form precipitation bands which merge with each other, whereas bands of heterogeneous antigens cross each other. This property permits determination of the homogeneity of the antigenic structure of various objects tested.
Components in the precipitation reaction in gel are agar gel, antigen, and antibodies. For the purpose of quality control of the precipitation test in gel, the test system comprised of known homologous antibodies and antigens is utilized.
The antigen used in a precipitation test should be concentrated, while the sera (from patients or immunized animals) should be of a high titre.
Procedure. To prepare gel, use 0.8-1 per cent solution of Difco’s agar or agarose on isotonic sodium chloride solution, which is layered on clean slides 1-
The precipitation test in gel is widely employed in the diagnosis of diseases caused by viruses, Rickettsia. and bacteria producing exotoxins. It has become of great practical significance with regard to determining the toxigenicity of Corynebacteria of diphtheria.
Immunoelectrophoresis (IEP) test allows analysis and identification of individual antigens in a multi-component system (Fig. 1, 3). The IEP test is based on the electrophoretic division of antigens in the gel with their subsequent precipitation by antibodies of the immune serum. To perform immunoelectrophoresis, glass plates with. an agar layer are used. First, the antigens placed in the centre of a plate are divided in the electrical field. Then, immune serum is added to an agar slit running parallel to the line of the antigen division. As a result of their mutual diffusion, the antigens and antibodies form the arches of precipitation at the place where they meet.
Counterimmunoelectrophoresis (CIEP) test is based on counter-diffusion in the electrical field of antigens and antibodies and the appearance of a visible precipitate inside a transparent gel. In agar or agarose gel cut the wells 2-
Students practical activities
Prepare participle bacterial antigen.
Daily culture of staphylococci to wash off 5 ml of a sterile isotonic saline and to transfer in centrifugin tube. Bacterial culture some times washing by isotonic saline from nutritious media after everyone centrifuge. Received suspension standardize under the turbidity standard 10. Then standard suspension, which contains 1 billion of bacterial cells in 1 ml to inactivate on water bath at 100° C within 1 hour and received antigene to check up on completeness inactivation. The absence of growth of test of an antigene outritious media testifies about sterility of an antigene.
Determination of immunoglobulins concentration in blood serum by radial immunodiffusion technique.
There is Difco’s agar with Ig-M-antiserum (antiserum containing only antibodies to the IgM) in the Petri’s dish. Using special device make the wells
Examination of T lymphocytes using Е (erythrocytic)–rosette formation (EF).
Mix in the tube 0.2 ml of previously prepared suspension of lymphocytes, 0.2 ml of sheep erythrocytes as a 0.3 % suspension in isotonic sodium chloride solution, 0.1 ml of 199 medium and 0.1 ml of bull serum. Centrifuge the mixture at 1000 rotations per min for 5 minutes. Place the tube into a 37 °C incubator for 30 min and for 20 min into refrigerator. Then carefully suspend the pellet and count up in Goryaev’s camera 100–200 lymphocytes. Determine a percentage of the cells, which connected three or more erythrocytes. Absolute quantity of T lymphocytes in 1 ml count up according the formula:
AQ = L x P x Р1, where
AQ – absolute quantity of T lymphocytes, L – quantity of leukocytes in 1 ml of blood, P – % of lymphocytes in blood formula, Р1 – % of erythrocytic rosette formation cells.
Examination of B-RFC in ready smears.
Count the B-RFC like in previous experiment according the same formula. Pay attention: for determination of B-RFC are used the erythrocytes of mice.
To carry out presumptive agglutination test on glass slides.
Presumptive agglutination test. A presumptive AT is performed on glass slides. Using a Pasteur pipette, transfer several drops of serum of low (1:10-1:20) dilutions and a drop of isotonic saline for control on a grease-free glass slide. Into each drop of the serum as well as in the control drop, inoculate a loopful of 24-hour living culture of the microorganism picked from the surface of a solid nutrient medium or pipette one drop of the suspension of dead microorganisms (diagnosticum). The inoculated culture is thoroughly mixed until the drop of liquid is uniformly turbid.
The reaction takes place at room temperature. Inspect visually the results in 5-10 min; occasionally one may use a 5 X magnifying lens for this purpose. If the glass slides are placed into a humid closed chamber to prevent evaporation, the results of the test may be read in 30-40 min as well.
A positive test is indicated by the appearance in the drop with serum of large or small flakes, readily visible upon rocking of the cover-slip. In a negative test, the fluid remains uniformly turbid.
Slide agglutination
In cases where the number of microorganisms is small and the results of the test are difficult to interpret, dry the drop of the inoculated serum, fix the preparation, stain it with Pfeifier’s fuchsine, and study under the microscope. In a positive test, a microscopic field is largely free of microorganisms but they are accumulated in some places. In a negative test, microorganisms are uniformly distributed throughout the microscopic field. This test is known as microagglutination.
To carry out agglutination test for serologic diagnostics of brucellosis
Schematic Representation of the Agglutination Reaction
|
Ingredient |
Number of the test tube |
|||||
|
1 |
2 |
3 |
4 |
5 antigen control |
6 serum control |
|
|
Isotonic sodium chloride solution, ml |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
– |
|
The patient’s serum in a 1: 50 dilution, ml |
1,0® |
1,0® |
1,0® |
1,0® |
– |
1,0 |
|
Dilution of the serum |
1:100 |
1:200 |
1:400 |
1:800 |
– |
1:50 |
|
Brucellar diagnosti- cum, drops |
2 |
2 |
2 |
2 |
2 |
2 |
|
Incubation at 37 |
||||||
The absence of agglutination in the control tubes and the presence of suspended flocculi in the tested tubes point to a positive test. Intensity of the reaction is denoted with pluses. In complete agglutination (++++), the liquid is completely transparent, while on the bottom of the test tube there is a floccular sediment of agglutinated microorganisms. The lesser the number of agglutinated microorganisms, the more turbid is the fluid and the smaller is the floccular pellet on the bottom (+++, ++, +). In a negative test (–), there is no sediment, the suspension remains uniformly turbid, showing no difference from the content of the test tube with the antigen control.
To carry out indirect hemagglutination test for serological diagnosis of typhoid fever .
Test results are assessed after complete erythrocyte sedimentation in control (6 well) –markedly localized erythrocytes sediment (“rouleaus”), In the experimental wells rapid erythrocytes agglutination with starlike, marginally festooned sediment (“umbrella”) on the bottom are observed. The titer of serum is its maximum dilution, which causes hemagglutination.
Schematic Representation of the indirect hemagglutination test
|
Ingredient |
Number of the lunula |
|||||
|
1 |
2 |
3 |
4 |
5 |
6 antigen control |
|
|
Isotonic sodium chloride solution, ml |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
|
Patient’s serum diluted 1: 50, ml |
1.0® |
1.0® |
1.0® |
1.0® |
1.0 |
– |
|
Obtained serum dilution |
1:100 |
1:200 |
1:400 |
1:800 |
1:1600 |
– |
|
Typhoid erythrocyte diagnosticum, ml |
0.25 |
0.25 |
0.25 |
0.25 |
0.25 |
0.25 |
|
Incubation at 37 °C for 2-3 hrs |
||||||
Indirect haemagglutinatyion test
To carry out ring precipitation test for determination of blood specificity.
Iarrow test-tube pour 0.9 ml of blood lysate and carefully, holding under an angle 450, with Pasteur pipette slowly layer on the wall (separately for everyone) 0.1 ml of diagnostic sera, which precipitated proteins of the man and different kinds of animals. The appearance of precipitate as the white ring on limit of two fluids the response is considered positive.
To carry out Askoli’s thermoprecipitation test.
The test is performed the same with previous, but only as a precipitinogen thermoextract from skin of animals is used, which carefully layer on 0,2–0,3 ml antianthracic serum. As a control isotonic sodium chloride solution, negative serum, standard antigen are used. The test should be positive with a standard and examined antigen.
To carry out immunodiffusion test (Uden technique).
Into a narrow test-tube pour melted transparent agar with an antiserum against protein of the man. After cooling the agar on its surface carefully slowly extract of human proteins is layered. At positive test precipitation lines appear in a gel.
To carry out double diffusion precipitation (Ouchterlony technique) test in gel.
In plastic dish pour gel and do a few holes. One lunula allocates in the center of dish another ones are near it (approximately 1-
Lysins and the Lysis Reaction. Lysins are specific antibodies which cause the dissolution of bacteria, plant and animal cells.
Under the influence of antibodies and a substance contained iormal serum, complement, the dissolution of microbial cells (bacteriolysis) takes place, or bactericidal action accompanied by destruction of microbes without any noticeable morphological changes occurs.
In 1884 V. Gromann established the bactericidal action of normal serum on the microbes of anthrax. V. Isaiev and R. Pfeiffer revealed antibodies (bacteriolysins) dissolving bacteria in the blood of immune animals. The Isaiev-Pfeiffer phenomenon may be reproduced in guinea pigs, actively or passively immunized against cholera. When a culture of cholera vibrios is injected into the peritoneal cavity of an immunized guinea pig, they lose their motility fairly rapidly, swell, become spherical, then granular and then finally completely disappear and dissolve. The same phenomenon is observed during simultaneous injection of live cholera vibrios and anticholera serum into a guinea pig. E. Metchnikoff and J. Bordet established that bacteriolysis may be observed outside the body by adding fresh immune serum to a bacterial suspension. In later investigations it was established that bacteriolysis depends not only on the antibody which appears under the influence of immunization, but also on the thermolabile substance (complement) found in all kinds of fresh serum, and which is disintegrated by heating at
The complement-fixation reaction has a high specificity and a marked sensitivity.
According to the mechanism of action this reaction is the most complex m comparison to reactions of agglutination and precipitation and proceeds m two phases. In the first phase precipitation occurs between the antigen and antibody (mutual adsorption), and in the second, fixation of the complement by the antibody-antigen complex takes place.
Complement participates in all immunological reactions, while in some reactions the presence of complement is obligatory (lysis, complement-fixation), in others it is non-obligatory (neutralization of toxin by antitoxin, precipitation, agglutination and opsonization).
The complement-fixation reaction is used in the diagnosis of glanders, syphilis (Wassermann reaction), etc. In recent years it has been used successfully in discerning typhus fever, Q fever and other rickettsioses and many virus diseases. Modifications of the complement fixation reaction have been devised for determining antibodies as well as antigens in the blood of patients. Preparation and titration of the ingredients and the method of the test are described in more detail in a manual.
Lysis Test. The term lysis reaction refers to dissolvement of the antigen conjugated with antibodies in the presence of a complement. Depending on the nature of antigens participating in the lysis reaction, it may be called spirochaetolysis, vibrionolysis, bacteriolysis, haemolysis, etc. Antibodies involved in the corresponding reactions are called spirochaetolysins, vibrionolysins, haemolysins, etc. Lysins exert their action only in the presence of a complement.
Most microorganisms with the exception of the cholera vibrio and Treponema are resistant to the lytic action of antibodies. Hence,. the lysis test has failed to find the wide-scale use in laboratory practice.
In carrying out the lysis reaction (Table 1), the immune serum is heated for 30 min at
Place the tubes into a
If the serum to be tested contains lysins, the number of colonies on a nutrient medium inoculated with the material to be assayed will be many times lower than in dishes containing the material from the control tubes.
The haemolysis reaction is used as an indicator system in the complement-fixation test.
The complement-fixation (CF) test belongs to complex serological reactions. It requires five ingredients to be performed; namely, an antigen, an antibody and complement (the first system), sheep red blood cells and haemolytic serum (the second system). Specific interaction of the antigen and antibody is attended by adsorption (binding) of the complement. Since the process of complement binding cannot be visualized, Bordet and Gangou have proposed to employ a haemolytic system (sheep erythrocytes plus haemolytic serum) as an indicator which shows whether the complement is fixed by the antigen-antibody complex. If the antigen and antibody correspond to each other, the complement is bound by this complex and no haemolysis takes place. If the antibody does not correspond to the antigen, the complex fails to be formed and free complement combines with the other system causing haemolysis.
CF, as other serological tests, may be used for identifying specific antibodies by a known antigen as well as for determining an antigen by known antibodies. The performance of the CF test calls for special preparation. The glassware (test tubes, pipettes, vials) are thoroughly washed and care is takeot to use them for other purposes. All ingredients of the reaction are prepared and titrated prior to the main test.
1. Before the test, the serum (either obtained from a patient or the diagnostic one) is heated on a water bath at
Some sera, particularly those from immunized animals, possess anticomplement properties, i.e., they can bind complement in the absence of a homologous antigen. This property of some sera is eliminated by treating them with carbon dioxide, heating at 57-58
2. Cultures of variable killed microorganisms, their lysates, bacterial components, of abnormal and normal organs, and tissue lipids, as well as viruses and virus-containing materials may be used as antigens for CF. Many antigens from microorganisms are available commercially.
3. The anti-complement activity of antigens is eliminated by such methods as thermolysis (multiple freezing and thawing), treatment with lipid solvents (ether, chloroform, acetone), and alcohols (methanol, ethanol).
Guinea pig serum collected immediately before a reaction is used as a complement; dry complement may also be employed. To obtain the basic solution for subsequent titration, the complement is diluted 1:10 with isotonic saline.
4. Sheep erythrocytes are employed as a 3 per cent suspension in isotonic sodium chloride solution. A blood sample (100-150 ml) is drawn from the jugular vein, put, into a sterile jar with glass beads, defibrinated by shaking for 10-15 min, and filtered through 3-4 layers of sterile gauze to remove fibrin. Erythrocytes are washed three times with isotonic saline by adding it to the erythrocytic sediment until the initial volume of blood is achieved. Red blood cells may be stored for 5-6 days at 4-6 “C. Their shelf life is increased if they are preserved by using either formalin (0.1 ml of undiluted formalin per 80 ml of defibrinated blood) or some other technique.
5. Haemolytic serum for complement-fixation is obtained in the following manner. Rabbits are immunized by injecting into the ear vein 50 per cent suspension of washed sheep erythrocytes (by 1-ml portions 4-6 times every other day). Seven days after the last injection the serum tested is obtained. If the serum titre is equal to or over 1:1200, bloodletting is performed. The serum is heated for 30min at 56
Titration of haemolytic serum. Serum is titrated by mixing 0.5 ml of the serum (in dilution 1:600; 1:1200; 1:1600; 1:3200, etc.) with 0.5 ml of 3 per cent suspension of red blood cells and 0.5 ml of fresh complement in dilution 1:10 (Table 2). The volume of the mixture for reaction in the control tubes is adjusted to 1.5 ml by adding 0.5 ml of isotonic sodium chloride solution. The results of the reaction are read after 1-hour incubation at
Complement titration. Before the test, the basic solution of complement (1:10) is dispensed into a series of test tubes in quantities varying from 0.05 to 0.5 ml, and then isotonic sodium chloride solution is added to each tube, bringing the volume of the fluid to 1.5 ml. The test tubes are incubated at
To perform the test, one takes the working dose of complement (contained in an 0.5-ml volume) exceeding the titre by 20-30 per cent.
Table 2
Schematic Representation of Haemolytic Serum Titration
|
Ingredient, ml |
Tube |
|||
|
1
|
2 |
3
|
4 |
|
|
test |
complement control |
haemolytic serum control |
erythrocyte control |
|
|
Haemolytic serum in dilutions 1:600, 1:1200, 1 :1600, 1 :3200, |
0.5 |
0.5 |
– |
– |
|
Sheep erythrocyte suspension |
0.5 |
0.5 |
0.5 |
0.5 |
|
Complement in 1 : 10 dilution |
0.5 |
– |
0.5 |
– |
|
Isotonic sodium chloride solution |
– |
0.5 |
0.5 |
1.0 |
|
Incubation at 37º C for 1 h |
||||
Table 3
Schematic Representation of Complement Titration
|
Ingredient, ml |
Number of the test tube |
|||||||||
|
Complement in 1:10 dilution |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
0.05 |
0.1 |
0.15 |
0.2 |
0.25 |
0.3 |
0.35 |
0.4 |
0.45 |
0.5 |
|
|
Isotonic sodium chloride solution |
1.45 |
1.4 |
1.35 |
1.3 |
1.25 |
1.2 |
1.15 |
1.1 |
1.05 |
1.0 |
|
Haemolytic system |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
1.0 |
|
Incubation at 37 ºC for 30 min |
||||||||||
Antigen titration. Antigens employed in complement fixation may adsorb a certain amount of complement, i.e., may have inherent anti-complement properties. For this reason antigens are titrated in the presence of the working dose of complement before the lest. Commercially available specific antigens have less marked anti-complement properties. Their titre is determined less frequently (for example, once a month in view of its possible reduction during storing). In this case various amounts of the basic dilution specified in the instruction are used.To determine the titre of the antigen, it is dispensed into a number of test tubes in decreasing amounts varying from 0.5 to 0.05 ml, bringing the volume to 1 ml by adding sodium chloride solution. Then, 0.5 ml of the working dose of complement is added to each tube and they are placed in
The titre of the antigen is its lowest amount sufficient to bring about complete haemolysis. For complement fixation the working dose of the antigen constituting about half to two-thirds of the titre is utilized. Antigens in whose presence the complement titre decreases by over 30 per cent are not suitable for the test.
Basic complement-fixation test. The total volume of ingredients involved in the reaction is 2.5 ml, the volume of the working dose of each of them is 0.5 ml. The diagram of the complement-fixation reaction shows (Table 4) that in the first test tube one introduces serum in the appropriate dilution, antigen and complement; into the second, serum in the appropriate dilution, complement and isotonic sodium chloride solution (serum control); into the third one, antigen, complement and isotonic saline (antigen control).
Table 4
Schematic Representation of the Basic CF Test
|
No of system |
Ingredient, ml
|
Number of the tube |
||
|
1
|
2 |
3
|
||
|
test |
serum control |
antigen control |
||
|
I |
Serum to be assayed in dilutions 1:5, 1:10, 1:20, 1:40, etc. |
0.5 |
0.5 |
– |
|
Antigen (working dose) |
0.5 |
– |
0.5 |
|
|
Complement (working dose) |
0,5 |
0,5 |
0,5 |
|
|
Isotonic sodium chloride solution |
– |
0,5 |
0,5 |
|
|
Incubation at 37 °C for 1 h |
||||
|
II |
Haemolytic system (haemolytic serum in triple titre + 3% suspension of sheep erythrocytes) |
1,0 |
1,0 |
1,0 |
|
Incubation at 37 ºC for 45 min |
||||
Simultaneously, a haemolytic system is prepared by mixing 2-ml portions of haemolytic serum in a triple titre (in relation to the one denoted in the instruction) and’ 3 per cent suspension of sheep erythrocytes (with regard to the initial volume of blood). The test tubes are incubated at 37 ‘C for 1 h, then 1 ml of the haemolytic system (the second system) is added to each of the first three tubes (the first system). After thoroughly mixing the ingredients the test tubes are reincubated at 37 °C for 1 h. The results of the reaction are read both preliminarily after removal of the tubes from the incubator and, finally, after they have stood for 15-18 hrs in a refrigerator or at room temperature.
In the final reading of the results the intensity of the reaction is expressed in pluses: (++++), a markedly positive reaction characterized by complete inhibition of haemolysis (the fluid in the tube is colourless, all red blood cells have settled on the bottom); (+++, ++), positive reaction manifested by the intensification of the liquid colour due to haemolysis and by a diminished number of red blood cells in the residue; (+), mildly positive reaction (the fluid is intensely colourful and there is only a small amount of erythrocytes collected on the bottom of the tube). If the reaction is negative (—), there is a complete haemolysis, and the fluid in the tube is intensely pink (varnish blood).
A number of complement-fixation modifications have been proposed, which are distinguished by elevated sensitivity and lesser volume of the ingredients used. Thus, the volume of ingredients for the CF test in the cold is 1 ml. For the drop CF test one takes 1 drop of serum + 1 drop of antigen +1 drop of complement + 2 drops of haemolytic system.
Despite its complexity, complement fixation is a sensitive and specific test, being used for these reasons for the diagnosis of many infectious diseases. Using the CF reaction, one can detect complement-binding antibodies in the blood serum obtained from patients with syphilis (Wassermann’s reaction), glanders, chronic gonorrhoea, rickettsiosis, viral diseases, etc.
Complement-binding antibodies make their appearance in the first days of the infection, yet their titre is relatively low. As a rule, antibodies reach the highest titre on the 7 th-10th-14 th day of the disease. Therefore, the most reliable are data obtained as a result of examining paired sera withdrawn at the onset of the disease and during convalescence.
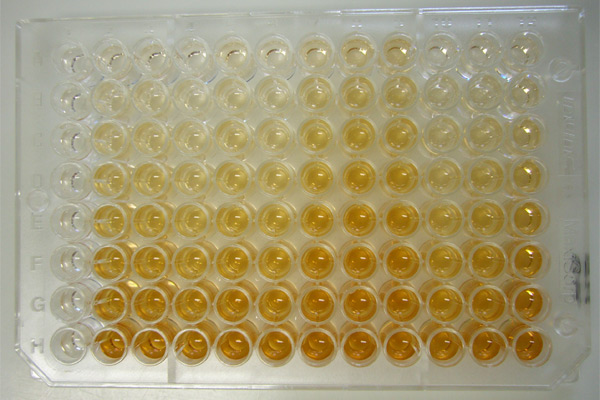
Enzyme-linked immunosorbent assay (ELISA)
History
Before the development of the ELISA, the only option for conducting an immunoassay was radioimmunoassay, a technique using radioactively labeled antigens or antibodies. In radioimmunoassay, the radioactivity provides the signal, which indicates whether a specific antigen or antibody is present in the sample. Radioimmunoassay was first described in a scientific paper by Rosalyn Sussman Yalow and Solomon Berson published in 1960.[1]
Because radioactivity poses a potential health threat, a safer alternative was sought. A suitable alternative to radioimmunoassay would substitute a nonradioactive signal in place of the radioactive signal. When enzymes (such as peroxidase) react with appropriate substrates (such as ABTS or 3,3’,5,5’-tetramethylbenzidine), a change in color occurs, which is used as a signal. However, the signal has to be associated with the presence of antibody or antigen, which is why the enzyme has to be linked to an appropriate antibody. This linking process was independently developed by Stratis Avrameas and G. B. Pierce.[2] Since it is necessary to remove any unbound antibody or antigen by washing, the antibody or antigen has to be fixed to the surface of the container; i.e., the immunosorbent must be prepared. A technique to accomplish this was published by Wide and Jerker Porath in 1966.[3]

Paramedic assistant prepares the analyses in ELISA laboratory
In 1971, Peter Perlmann and Eva Engvall at Stockholm University in Sweden, and Anton Schuurs and Bauke van Weemen in the Netherlands independently published papers that synthesized this knowledge into methods to perform EIA/ELISA.[4][5]
Traditional ELISA typically involves chromogenic reporters and substrates that produce some kind of observable color change to indicate the presence of antigen or analyte. Newer ELISA-like techniques use fluorogenic, electrochemiluminescent, and real-time PCR reporters to create quantifiable signals. These new reporters can have various advantages, including higher sensitivities and multiplexing.[6][7] In technical terms, newer assays of this type are not strictly ELISAs, as they are not “enzyme-linked”, but are instead linked to some nonenzymatic reporter. However, given that the general principles in these assays are largely similar, they are often grouped in the same category as ELISAs.
In 2012 an ultrasensitive, enzyme-based ELISA test using nanoparticles as a chromogenic reporter was able to give a naked-eye colour signal from the detection of mere attograms of analyte.[8]
Enzyme-linked immunosorbent assay (ELISA) is a test that uses antibodies and color change to identify a substance.
ELISA is a popular format of a “wet-lab” type analytic biochemistry assay that uses a solid-phase enzyme immunoassay (EIA) to detect the presence of a substance, usually an antigen, in a liquid sample or wet sample.
The ELISA has been used as a diagnostic tool in medicine and plant pathology, as well as a quality-control check in various industries.
Antigens from the sample are attached to a surface. Then, a further specific antibody is applied over the surface so it can bind to the antigen. This antibody is linked to an enzyme, and, in the final step, a substance containing the enzyme’s substrate is added. The subsequent reaction produces a detectable signal, most commonly a color change in the substrate.
Performing an ELISA involves at least one antibody with specificity for a particular antigen. The sample with an unknown amount of antigen is immobilized on a solid support (usually a polystyrene microtiter plate) either non-specifically (via adsorption to the surface) or specifically (via capture by another antibody specific to the same antigen, in a “sandwich” ELISA). After the antigen is immobilized, the detection antibody is added, forming a complex with the antigen. The detection antibody can be covalently linked to an enzyme, or can itself be detected by a secondary antibody that is linked to an enzyme through bioconjugation. Between each step, the plate is typically washed with a mild detergent solution to remove any proteins or antibodies that are not specifically bound. After the final wash step, the plate is developed by adding an enzymatic substrate to produce a visible signal, which indicates the quantity of antigen in the sample.
Of note, ELISA can perform other forms of ligand binding assays instead of strictly “immuno” assays, though the name carried the original “immuno” because of the common use and history of development of this method. The technique essentially requires any ligating reagent that can be immobilized on the solid phase along with a detection reagent that will bind specifically and use an enzyme to generate a signal that can be properly quantified. In between the washes, only the ligand and its specific binding counterparts remain specifically bound or “immunosorbed” by antigen-antibody interactions to the solid phase, while the nonspecific or unbound components are washed away. Unlike other spectrophotometric wet lab assay formats where the same reaction well (e.g. a cuvette) can be reused after washing, the ELISA plates have the reaction products immunosorbed on the solid phase which is part of the plate, so are not easily reusable.
Principle
As a “wet lab” analytic biochemistry assay, ELISA involves detection of an “analyte” (i.e. the specific substance whose presence is being quantitatively or qualitatively analyzed) in a liquid sample by a method that continues to use liquid reagents during the “analysis” (i.e. controlled sequence of biochemical reactions that will generate a signal which can be easily quantified and interpreted as a measure of the amount of analyte in the sample) that stays liquid and remains inside a reaction chamber or well needed to keep the reactants contained; It is opposed to “dry lab” that can use dry strips – and even if the sample is liquid (e.g. a measured small drop), the final detection step in “dry” analysis involves reading of a dried strip by methods such as reflectometry and does not need a reaction containment chamber to prevent spillover or mixing between samples.
As a heterogenous assay, ELISA separates some component of the analytical reaction mixture by adsorbing certain components onto a solid phase which is physically immobilized. In ELISA, a liquid sample is added onto a stationary solid phase with special binding properties and is followed by multiple liquid reagents that are sequentially added, incubated and washed followed by some optical change (e.g. color development by the product of an enzymatic reaction) in the final liquid in the well from which the quantity of the analyte is measured. The qualitative “reading” usually based on detection of intensity of transmitted light by spectrophotometry, which involves quantitation of transmission of some specific wavelength of light through the liquid (as well as the transparent bottom of the well in the multiple-well plate format). The sensitivity of detection depends on amplification of the signal during the analytic reactions. Since enzyme reactions are very well known amplification processes, the signal is generated by enzymes which are linked to the detection reagents in fixed proportions to allow accurate quantification – thus the name “enzyme linked”.
The analyte is also called the ligand because it will specifically bind or ligate to a detection reagent, thus ELISA falls under the bigger category of ligand binding assays. The ligand-specific binding reagent is “immobilized”, i.e., usually coated and dried onto the transparent bottom and sometimes also side wall of a well (the stationary “solid phase’/”solid substrate” here as opposed to solid microparticle/beads that can be washed away), which is usually constructed as a multiple-well plate known as the “ELISA plate”. Conventionally, like other forms of immunoassays, the specificity of antigen–antibody type reaction is used because it is easy to raise an antibody specifically against an antigen in bulk as a reagent. Alternatively, if the analyte itself is an antibody, its target antigen can be used as the binding reagent.
Types
“Indirect” ELISA

Direct ELISA diagram
The steps of “indirect” ELISA follows the mechanism below:-
- A buffered solution of the antigen to be tested for is added to each well of a microtiter plate, where it is given time to adhere to the plastic through charge interactions.
- A solution of nonreacting protein, such as bovine serum albumin or casein, is added to block any plastic surface in the well that remains uncoated by the antigen.
- The primary antibody is added, which binds specifically to the test antigen coating the well. This primary antibody could also be in the serum of a donor to be tested for reactivity towards the antigen.
- A secondary antibody is added, which will bind the primary antibody. This secondary antibody often has an enzyme attached to it, which has a negligible effect on the binding properties of the antibody. In other cases, as in the diagram to the left, the primary antibody itself is conjugated to the enzyme.
- A substrate for this enzyme is then added. Often, this substrate changes color upon reaction with the enzyme. The color change shows the secondary antibody has bound to primary antibody, which strongly implies the donor has had an immune reaction to the test antigen. This can be helpful in a clinical setting, and in research.
- The higher the concentration of the primary antibody present in the serum, the stronger the color change. Often, a spectrometer is used to give quantitative values for color strength.
The enzyme acts as an amplifier; even if only few enzyme-linked antibodies remain bound, the enzyme molecules will produce many signal molecules. Within common-sense limitations, the enzyme can go on producing color indefinitely, but the more primary antibody is present in the donor serum, the more secondary antibody + enzyme will bind, and the faster the color will develop. A major disadvantage of the indirect ELISA is the method of antigen immobilization is not specific; when serum is used as the source of test antigen, all proteins in the sample may stick to the microtiter plate well, so small concentrations of analyte in serum must compete with other serum proteins when binding to the well surface. The sandwich or direct ELISA provides a solution to this problem, by using a “capture” antibody specific for the test antigen to pull it out of the serum’s molecular mixture.
ELISA may be run in a qualitative or quantitative format. Qualitative results provide a simple positive or negative result (yes or no) for a sample. The cutoff between positive and negative is determined by the analyst and may be statistical. Two or three times the standard deviation (error inherent in a test) is often used to distinguish positive from negative samples. In quantitative ELISA, the optical density (OD) of the sample is compared to a standard curve, which is typically a serial dilution of a known-concentration solution of the target molecule. For example, if a test sample returns an OD of 1.0, the point on the standard curve that gave OD = 1.0 must be of the same analyte concentration as the sample.
Sandwich ELISA

A sandwich ELISA. (1) Plate is coated with a capture antibody; (2) sample is added, and any antigen present binds to capture antibody; (3) detecting antibody is added, and binds to antigen; (4) enzyme-linked secondary antibody is added, and binds to detecting antibody; (5) substrate is added, and is converted by enzyme to detectable form.
A less-common variant of this technique, a “sandwich” ELISA, is used to detect sample antigen. The steps are:
1. A surface is prepared to which a known quantity of capture antibody is bound.
2. Any nonspecific binding sites on the surface are blocked.
3. The antigen-containing sample is applied to the plate.
4. The plate is washed to remove unbound antigen.
5. A specific antibody is added, and binds to antigen (hence the ‘sandwich’: the Ag is stuck between two antibodies)
6. Enzyme-linked secondary antibodies are applied as detection antibodies that also bind specifically to the antibody’s Fc region (nonspecific).
7. The plate is washed to remove the unbound antibody-enzyme conjugates.
8. A chemical is added to be converted by the enzyme into a color or fluorescent or electrochemical signal.
9. The absorbency or fluorescence or electrochemical signal (e.g., current) of the plate wells is measured to determine the presence and quantity of antigen.
The image to the right includes the use of a secondary antibody conjugated to an enzyme, though, in the technical sense, this is not necessary if the primary antibody is conjugated to an enzyme. However, use of a secondary-antibody conjugate avoids the expensive process of creating enzyme-linked antibodies for every antigen one might want to detect. By using an enzyme-linked antibody that binds the Fc region of other antibodies, this same enzyme-linked antibody can be used in a variety of situations. Without the first layer of “capture” antibody, any proteins in the sample (including serum proteins) may competitively adsorb to the plate surface, lowering the quantity of antigen immobilized. Use of the purified specific antibody to attach the antigen to the plastic eliminates a need to purify the antigen from complicated mixtures before the measurement, simplifying the assay, and increasing the specificity and the sensitivity of the assay.
A descriptive animation of the application of sandwich ELISA to home pregnancy testing can be found here.
Competitive ELISA
A third use of ELISA is through competitive binding. The steps for this ELISA are somewhat different from the first two examples:
1. Unlabeled antibody is incubated in the presence of its antigen (sample).
2. These bound antibody/antigen complexes are then added to an antigen-coated well.
3. The plate is washed, so unbound antibody is removed. (The more antigen in the sample, the less antibody will be able to bind to the antigen in the well, hence “competition”.)
4. The secondary antibody, specific to the primary antibody, is added. This second antibody is coupled to the enzyme.
5. A substrate is added, and remaining enzymes elicit a chromogenic or fluorescent signal.
6. The reaction is stopped to prevent eventual saturation of the signal.
Some competitive ELISA kits include enzyme-linked antigen rather than enzyme-linked antibody. The labeled antigen competes for primary antibody binding sites with the sample antigen (unlabeled). The more antigen in the sample, the more labeled antigen is retained in the well and the stronger the signal.
Commonly, the antigen is not first positioned in the well.
For the detection of HIV antibodies, the wells of microtiter plate are coated with the HIV antigen. Two specific antibodies are used, one conjugated with enzyme and the other present in serum (if serum is positive for the antibody). Cumulative competition occurs between the two antibodies for the same antigen, causing a stronger signal to be seen. Sera to be tested are added to these wells and incubated at 37°C, and then washed. If antibodies are present, the antigen-antibody reaction occurs. No antigen is left for the enzyme-labelled specific HIV antibodies. These antibodies remain free upon addition and are washed off during washing. Substrate is added, but there is no enzyme to act on it, so positive result shows no color change.
Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool for determining serum antibody concentrations (such as with the HIV test or West Nile virus). It has also found applications in the food industry in detecting potential food allergens, such as milk, peanuts, walnuts, almonds, and eggs.[10] ELISA can also be used in toxicology as a rapid presumptive screen for certain classes of drugs.

![]()
Enzyme-linked immunosorbent assay plate
The ELISA was the first screening test widely used for HIV because of its high sensitivity. In an ELISA, a person’s serum is diluted 400 times and applied to a plate to which HIV antigens are attached. If antibodies to HIV are present in the serum, they may bind to these HIV antigens. The plate is then washed to remove all other components of the serum. A specially prepared “secondary antibody” — an antibody that binds to other antibodies — is then applied to the plate, followed by another wash. This secondary antibody is chemically linked in advance to an enzyme.
Thus, the plate will contain enzyme in proportion to the amount of secondary antibody bound to the plate. A substrate for the enzyme is applied, and catalysis by the enzyme leads to a change in color or fluorescence. ELISA results are reported as a number; the most controversial aspect of this test is determining the “cut-off” point between a positive and a negative result.
A cut-off point may be determined by comparing it with a known standard. If an ELISA test is used for drug screening at workplace, a cut-off concentration, 50 ng/ml, for example, is established, and a sample containing the standard concentration of analyte will be prepared. Unknowns that generate a stronger signal than the known sample are “positive.” Those that generate weaker signal are “negative”.
Dr Dennis E Bidwell and Alister Voller created the ELISA test to detect various kind of diseases, such as malaria, Chagas disease, and Johne’s disease.[11] ELISA tests also are used as in in vitro diagnostics in medical laboratories. The other uses of ELISA include:
- detection of Mycobacterium antibodies in tuberculosis
- detection of rotavirus in feces
- detection of hepatitis B markers in serum
- detection of enterotoxin of E. coli in feces
- detection of HIV antibodies in blood samples