Criteria for diagnosis and differential programs of treatment of diffuse diseases of lungs.
COPD, CHRONIC BRONCHITIS
Chronic bronchitis is probably the most common debilitating respiratory disease in the
Clinical Findings
A. Symptoms and Signs: The hallmark of chronic bronchitis is chronic cough and sputum production. Specifically, the definition of chronic bronchitis requires that productive cough be present on most days for a minimum of 3 months in the year in at least 2 consecutive years in order to make the diagnosis. The disease is most commonly seen in smokers over age 35. Initially, cough with sputum production, often in the morning, may be the only symptom. Gradually the cough and sputum production increase and symptoms of dyspnea on exertion develop.
As the disease progresses, the patient’s course is usually marked by recurrent episodes of acute respiratory failure resulting from infectious exacerbations of the bronchitis. Clinically, these are marked by increased cough, change in sputum from clear and mucoid to purulent, fever, dyspnea, and varying degrees of respiratory distress. Respiratory failure often ensues, with both elevated Paco2 and diminished PaO2. These episodes are often reversible at first with appropriate antibiotics, bronchodilators, and respiratory therapy.
Signs and symptoms of cor pulmonale are frequent in chronic bronchitis and are exacerbated along with episodes of acute respiratory failure. Also is very important to differenciate Chronic Bronchitis and asthma (picture 1).

Picture 1 Quick guide to the differences between COPD and asthma
The course of the disease is one of gradual increase in frequency and severity of episodes of acute infection and respiratory failure, eventually resulting in intubation and the need for almost constant ventilatory assistance. Death usually occurs during an episode of respiratory failure.
Depending on the stage in which the patient is examined, the physical findings may vary. During relatively quiescent periods, the only finding may be increased anteroposterior diameter of the chest, hyperresonance to percussion, prolonged expiratory phase, scattered diffuse coarse to medium rhonchi and rales, and wheezing. Later, the patient may manifest the signs and symptoms of pulmonary hypertension and right ventricular failure, ie, increased second heart sound, pedal edema, hepatomegaly, and ascites. These patients commonly have a plethoric appearance resulting from secondary polycythemia.
If examined during an acute attack, the patient will be in respiratory distress, as evidenced by tachypnea and use of accessory muscles of respiration, in addition to the signs described above. Cough is often prominent, and cyanosis during acute attacks is not uncommon. For these reasons, patients with predominant bronchitis have been called “blue bloaters”.
B.: Routine laboratory studies often demonstrate the presence of secondary polycythemia (eg, hematocrit greater than 55%). Also, if right ventricular failure and hepatic congestion are present, there may be aberrations in liver function tests (SGOT, alkaline phosphatase, bilirubm, etc). Culture of sputum often shows H. influenzae and Streptococcus pneumoniae Several studies have indicated that these organisms may colonize the upper and lower airways of patients with chronic bronchitis, and their significance in the pathogenesis of acute exacerbations is unknown at present. Arterial blood gases show hypoxemia with either low, normal, or elevated Paco2.
If the patient has chronically retained CO2, serum bicarbonate will also be elevated, and there will be only a modest degree of acidemia. If C02 retention is the result of an acute exacerbation, there will not be an elevation of bicarbonate, and respiratory acidosis with acidemia may be pronounced
Pulmonary function studies uniformly show expiratory air flow obstruction. Vital capacity may be normal or reduced, and there is often evidence of air trapping, with increased residual volume, total lung capacity, and ratio of RV to TLC. Expirator reserve volume also is often diminished, and functional residual capacity is increased. Diffusion capacity for carbon monoxide may be normal or decreased.
More sophisticated pulmonary function studies show normal elastic recoil, increased airway resistance, and relatively normal compliance in pure bronchitis.
C. X-Ray Findings: Chest x-rays show (picture 2) evidence of pulmonary overinflation, with increased anteropostenor diameter, flattened diaphragms, and increased retrosternal air space (unless right ventricular hypertrophy has supervened). There are often prominent and increased bronchial markings at the lung bases, seen as parallel or tapering shadows (“tram lines”), which reflect the increased thickness of the bronchial wall. Bullae of varying sizes may be seen primarily in the upper lung fields rather than the lung bases (as m alpha-antitrypsin deficiency).
Picture 2. These radiographs of a patient with chronic obstructive pulmonary disease (COPD) reveal pulmonary hyperinflation. In the PA projection above the diaphragms are at the level of the eleventh posterior ribs and appear flat. The lateral radiograph below demonstrates the prominence of the anterior clear-space and of the AP diameter of the chest as well as the flat diaphragms.
Treatment
There is no doubt that smoking hastens the progression of emphysema and shortens the life span of patients with the disease. Patients must be vigorously encouraged to discontinue cigarette smoking and avoid exposure to other toxic inhalants when possible. Since the risk of infection resulting in respiratory failure is great, these patients should be immunized against pneumococci and influenza. The efficacy of prophylactic or regular antibiotic treatment is unproved and controversial, and it is reasonable to reserve antibiotics for management of acute exacerbations, as manifested by change in sputum character, increased dyspnea, etc.
The preferred drug is ampicillin, 250-500 mg every 6 hours, or erythromycin, 500 mg every 6 hours, for 5-7 days. When exacerbations are associated with respiratory failure, appropriate treatment measures must be instituted. Since many of these patients display varying degrees of chronic bronchospasm, most authorities treat chronically with bronchodilators. Oral aminophylline in dosages sufficient to give serum theophylime levels of 10-20 /mg/mL is the drug of choice (usual dosage 250-500 mg 4 times daily or 200 mg of sustained-release preparations twice daily). Inhaled bronchodilators may also be useful in the chronic management of these patients. The usual dosage is 2-3 puffs 4 times daily. Sympathomimetic beta-agonists (terbutaline, 2.5-5 mg 3 times daily, or metaproterenol, 10-20 mg 3 times daily) are often useful in chronic management.
Many patients with chronic bronchitis develop right ventricular failure, and there is also a considerable body of evidence suggesting that left ventricular dysfunction is common as well. For this reason, signs and symptoms referable to cardiac dysfunction (either rightor left-sided) are managed with diuretics. The administration of digitalis is controversial; some authors prefer not to use it unless there is documented evidence of left ventricular failure (ie, increased pulmonary capillary wedge pressure) or a digitalisresponsive arrhythmia (eg, atrial flutter).
The use of corticosteroids in patients with COPD/bronchitis is controversial also. Many authorities feel that there is a significant subgroup of patients who respond favorably to the administration of corticosteroids (eg, prednisone, 20-50 mg daily). No clinical or laboratory markers for this group of patients have been definitely identified, and there are no well-documented studies proving the efficacy of corticosteroid therapy. Nonetheless, many authorities use corticosteroids as a last resort in patients with severe disease.
Other measures, such as low-flow home oxygen and patient education regarding coughing and postural drainage exercises, are often useful. Treatment according to the stage is presented in piucture 3.
Picture 3 Therapy at each stage of COPD
Course & Prognosis
The course of the disease in any one patient is quite variable. However, in general there is progressive deterioration of pulmonary function with increasing frequency of respiratory failure episodes until patients with impaired respiratory or cardiac function or in patients debilitated by other illnesses. Treatment is with procaine penicillin, 1.2 million units intramuscularly daily, or one of the following given orally every 6 hours: penicillin G, 250-500 mg; tetracycline, 250-500 mg; ampicillin, 250-500 mg.
BRONCHIECTASIS
Essentials of Diagnosis:
• Chronic cough with expectoration of large amounts of purulent sputum; hemoptysis.
• Rales and rhonchi over lower lobes.
• X-ray of chest reveals little; bronchograms show characteristic dilatations.
General Considerations
Bronchiectasis is a dilatation of small and medium-sized bronchi resulting from destruction of bronchial elastic and muscular elements. It may be, caused by pulmonary infections (eg, pneumonia, pertussis, tuberculosis) or by a bronchial obstruction (eg, foreign bodies or extrinsic pressure). In many patients, a history of onset following one or more episodes of pulmonary infection, usually in early childhood, is obtained. However,since infection does not regularly produce significant bronchiectasis, unknown intrinsic host factors presumably are present. The incidence of
the disease has been reduced by treating pulmonary infections with antibiotics.
Clinical Findings
A. Symptoms and Signs: Most patients with bronchiectasis have a history of chronic cough with expectoration of large volumes of sputum, especially upon awakening. The sputum has a characteristic quality of “layering out” into 3 layers upon standing, a frothy top layer, a middle clear layer, and a dense particulate bottom layer. It is usually purulent in appearance and foul-smelling.
Intermittent
hemoptysis, occasionally in dangerous proportions, is often combined with intercurrent respiratory infections. Symptoms occur most often in patients with idiopathic bronchiectasis (ie, childhood respiratory infections). However, patients who have bronchiectasis secondary either to tuberculosis or chronic obstruction may not exhibit characteristic symptoms. Idiopathic bronchiectasis occurs most frequently in the middle and lower lobes and posttuberculous bronchiectasis in the upper lobes.
Hemoptysis is thought to result from erosion of bronchiolar mucosa with resultant destruction of underlying blood vessels. Pulmonary insufficiency may result from progressive destruction of pulmonary tissue.
Physical findings consist primarily of rales and rhonchi over the affected segments. If the condition is far-advanced, emaciation, cyanosis, and digital clubbing may appear.
B. Laboratory Findings: There are no characteristic laboratory findings. If hypoxemia is chronic and severe, secondary polycythemia may develop. There may be either restrictive or obstructive pulmonary function defects associated with bronchiectasis. Hypoxemia and hypocapnia or hypercapnia may also be associated with the disease, depending on the severity of the underlying condition. .
C.X-Ray Findings (figure 1-6): Plain films of the chest often show increased bronchopulmonary markings in affected segments; in severe cases there may be areas of radiodensities surrounding portions of radiolucency. Early in the course of bronchiectasis, however, the chest x-ray may be normal.
Iodized contrast media instilled into the bronchial tree (a bronchogram) demonstrates saccular, cylindric, or fusiform dilatation of small and medium bronchi with consequent loss of the normal branching pattern. Cylindric changes of bronchiectasis that may result from acute pneumonia will revert to normal after 6-8 weeks, but saccular dilatations represent long-standing damage and permanent disease.
Figure 1. PA (1A) and Lateral chest (1B) radiographs in a 64 year old lady with a chronic cough secondary to MAC. There is air space opacity in the middle lobe (solid arrows) and lingula (dashed arrows)
Figure 2. 83 year old lady with a chronic cough. CT scan at the level of the pulmonary artery demonstrates bronchiectasis (solid arrow)and tree-in-bud nodules (dashed arrows) involving the middle lobe, lingula and lower lobes, secondary to MAC
Figure 3. 83 year old lady with a chronic cough. Coronal reformat CT scan through the airways demonstrates extensive bronchiectasis, tree-in-bud nodules (dashed arrow) and right upper lobe segmental atelectasis (solid arrow) secondary to MAC
Figure 4. Bronchiectasis Radiograph Endobronchial radiographic dye is used to demonstrate the dilated bronchi in bronchiectasis.
Figure 5: Bronchogram showing extensive bronchiectasis of left lung
Figure 6: CT scan showing extensive bronchiectasis of left lung.
Differential Diagnosis
The differential diagnosis includes other disorders that lead to chronic cough, sputum production, and hemoptysis, ie, chronic bronchitis, tuberculosis, and bronchogenic carcinoma. The diagnosis of bronchiectasis is suggested by the patient’s history and can be confirmed only by bronchographic examination or histopathologic examination of surgically removed tissue.
Complications
Recurrent infection in poorly drained pulmonary segments leads to chronic suppuration and may cause pulmonary insufficiency. Complications include hemoptysis, respiratory failure, chronic cor pulmonale, and amyloidosis. There is also an increased incidence of brain abscess, which is thought to be secondary to abnormal anastomoses between bronchial (systemic) and pulmonary venous circulation. These anastomoses produce right-to-left shunts and allow for the dissemination of septic emboli.
Treatment
. A. General Measures and Medical Treatment:
1. Environmental changes– The patient should avoid exposure to all common pulmonary irritants such as smoke, fumes, and dust and should stop smoking cigarettes.
2. Control of bronchial secretions (improved drainage)–
a. Postural drainage often gives effective relief of symptoms and should be utilized in every case. The patient should assume the position that gives maximum drainage, usually lying on a bed in the prone, supine, or right or left lateral decubitus position with the hips elevated on several pillows and no pillow under the head. Any effective position should be maintained for 10 minutes, 2-4 times a day. The first drainage should be done upon awakening and tee last drainage at bedtime. Family members can be trained in the art of chest percussion to facilitate drainage of secretions.
b. Liquefaction of thick sputum may be promoted by inhaling warm mists and, in some cases, mucolytic agents such as acetylcysteine or 5% sodium bicarbonate given by aerosol may also be helpful.
3. Control of respiratory infection-Exposure to respiratory infections should be minimized and the patient should be vaccinated against influenza and pneumococcal pneumonia. Antibiotic therapy is indicated for acute exacerbations (ie, increased production of purulent sputum, hemoptysis, etc). Long-term or prophylactic antibiotic therapy is controversial, since it has not been conclusively shown to be of lasting benefit. Therefore, it seems rational to treat acute exacerbations in order to control infection but minimize the emergence of resistant strains. Because the bacteria most commonly involved are H inftuenzae and S pneumoniae, the drug most commonly employed is ampicillin, 250-500 mg orally every 6 hours for 5 days. Alternative therapies for the penicillin-allergic patient are erythromycin, given in the same dosage schedule as ampicillin, or trimethoprim-sulfamethoxazole, 2 double-strength tablets twice a day for 5 days.
B. Surgical Treatment: Surgical treatment is most often employed when hemoptysis with bron-chiectasis is recurrent and severe. Despite antibiotic therapy, localized bronchiectasis (eg, in a lower lobe or segment) with progressive uncontrolled infection and sputum production may be an indication for surgical removal of the affected segments.
Other Considerations
Bronchiectasis is also associated with mucoviscidosis. It is thought to be secondary to the thick viscid secretions that cannot be cleared by normal cough mechanisms and that lead to stasis of. sputum and chronic infection. This disorder, usually associated with sinusitis, may be accompanied by other manifestations of mucoviscidosis. Its most common organisms are S aureus or Pseudomonas aeruginosa.
Bronchiectasis is also associated with certain abnormalities of cellular ciliary function, the most common of which is Kartagener’s syndrome, a combination of sinusitis, situs in versus, and bronchiectasis. Patients with this disorder show immotile cilia secondary to ultrastructural abnormalities, stasis of sputum, failure to clear secretions, and chronic pulmonary infection that results in bronchiectasis.
Antibiotic treatment of mucoviscidosis and Kartagener’s syndrome must be guided by sensitivity studies of organisms cultured from sputum.
OCCUPATIONAL LUNG DISEASES
Picture 4-5. Hazardous ccupational environment
Lung disorders directly related to the inhalation of dust, fumes, vapors, and gases from the occupational environment (picture 4-5).
The effects of an inhaled agent depend on a number of factors—its physical properties, its chemical properties, and the susceptibility of the exposed person (see table 1). A particle is a solid particulate, a mist is a liquid particulate, a vapor is the gaseous form of substance that is normally a liquid, and gas is that physical state in which a substance has no fixed volume. In some instances, the inhaled material is deposited and retained in the lungs; if soluble, it is absorbed into the bloodstream. Insoluble particles and mists for the most part are removed by the body’s defenses.
The physical state of the inhaled agent is of great importance. Particles are deposited in the respiratory tract as the result of 3 physical processes: sedimentation, inertial impaction, and diffusion. Sedimentation depends on Stokes’ law, and is dependent on the particle’s density and the square of its diameter. Inertial impaction occurs in bifurcating airways when the momentum of a particle is sufficient to carry it along in its original path so that it impinges on the bronchial wall. Diffusion is due to kinetic energy present in all small particles that causes them to move at random. Larger particles between 6 and 25 M are deposited by sedimentation in the nose, and to a lesser extent in the conducting airways. Since they are too large to find their way into the lung parenchyma, they are known as the nonrespirable fraction. Particles of between 0.5 and 6 M are known as the respirable fraction and are most prone to be deposited in the gas-exchanging portions of the lung. Particles between 1 and 2 M are most often involved in the development of pneumoconiosis. Very small particles below 0.1 M are deposited in the lung parenchyma due to diffusion. Other physical properties also influence the effects of deposited particles. Thus, those asbestos fibers with the greatest penetrability are more likely to migrate to the pleura and cause mesothelioma. The chemical properties of the inhaled agent are also important. While quartz is markedly fibrogenic, particles of coal, carbon, and tin oxide are far less so.
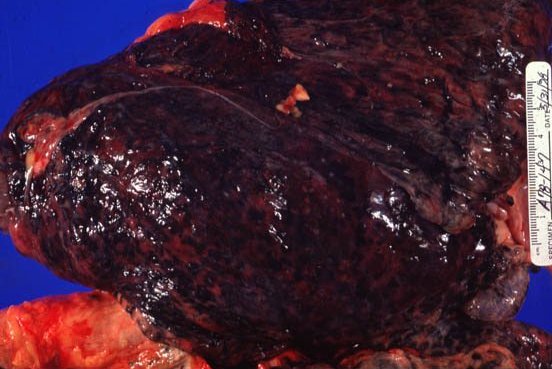
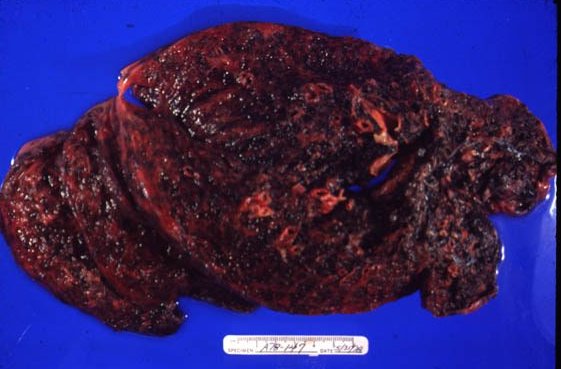
Picture 4. This is a lung from a coal miner.
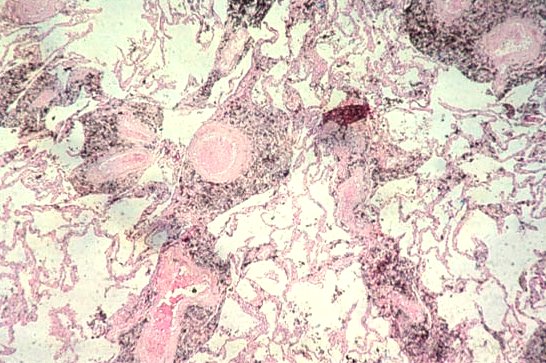

Picture 5. This is a polarized light view.
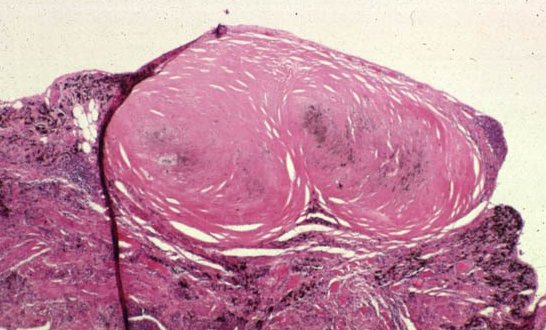
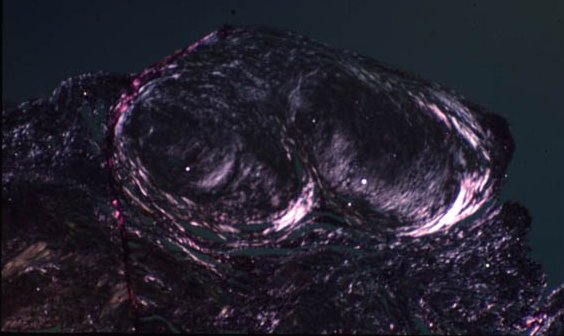
Picture 6. This miner’s lung has a preponderance of silicotic lesions.
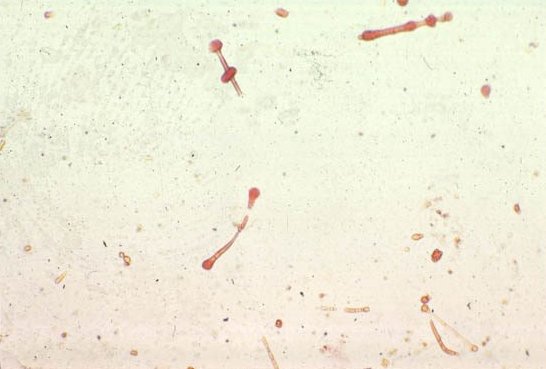
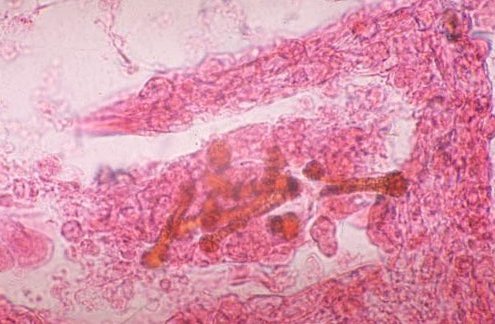

Picture 7. Here are asbestos fibers. The last slide shows a pleural plaque.
Individual susceptibility affects the development of occupational lung diseases, and there are marked variations in the rate of clearing of particles from the respiratory tract of different persons. Particles deposited in the dead space are removed by the mucociliary escalator more rapidly in some persons than in others;
the clearance rate is genetically determined. Particles deposited in the lung parenchyma are taken up by alveolar macrophages, and the latter migrate with their engulfed particles to the terminal bronchioles where they catch the mucociliary escalator, or into the interstitium of the lungs where they are transported to the lymph nodes.
The site of deposition of the particle is of prime importance and to a large extent governs the lung’s response (see table 2). Deposition of particles in the nose may lead to hay fever, which may be regarded as occupationally related in an agricultural worker. Septal perforation may be seen in chrome workers, and nasal cancer in furniture workers. Deposition of particles in the trachea and bronchi may induce 3 responses. First, there may be bronchoconstriction, which can result from an antigen-antibody reaction; e.g., as in some forms of occupational asthma. Second, as in byssinosis, the deposition of particles may (through pharmacologic mechanisms) cause the mast cells of the airways to produce bronchoconstrictors such as histamine and SRS-A. Next, the long-continued deposition of particles may induce mucus gland hypertrophy or bronchitis, which, in some instances,
table 1 . FACTORS INFLUENCING EFFECTS OF INHALED AGENTS
|
Physical properties Chemical properties Individual susceptibility |
Physical state (i.e., particle, mist, or gas; solubility, shape, density, penetrability, concentration, radioactivity) Acidity, alkalinity, fibrogenlclty, antigenicity Integrity of body’s defenses, immunologic status; i.e., atopy, tissue type (HLA type), airways geometry |
table 2. INFLUENCE OF SITE OF DEPOSITION OF THE RESPIRATORY TRACT
|
Site of Deposition |
Clinical Response |
|
Nose Trachea and bronchi Lung parenchyma |
Rhinitis, hay fever, septal perforation, nasal cancer Bronchoconstnction Antigen-antibody-mediated Pharmacologtcally induced Bronchitis Nonspecific response to inert dusts Lung cancer (radioactive dusts and gases) Extrinsic allergic alveolitis (organic dusts) Pneumoconiosis (mineral dusts) Acute pulmonary damage, bronchiolitis, pulmonary edema |
leads to a minor degree of chronic airflow obstruction. Finally, the deposition of asbestos fibers, and of dusts with radon daughters adsorbed to them, may lead to the development of lung cancer.
If particles deposited in the lung parenchyma are organic and antigenic, they may lead to the development of extrinsic allergic alveolitis (hypersensitivity pneumonia), an acute granulomatous process involving the alveoli and respiratory bronchioles. When the particle is inorganic, a fibrotic response may be seen that can be either focal and nodular as in typical silicosis, or diffuse and generalized as in asbestosis and berylliosis. Should the particle be inert (e.g., tin oxide), a benign pneumoconiosis without fibrosis develops. The inhalation of certain gases and vapors (e.g., mercury, cadmium, nitrogen dioxide) can cause acute pulmonary edema, acute alveolitis, and bronchiolitis obliterans.
DISEASES DUE TO INORGANIC (MINERAL) DUSTS
FIBROGENIC DUST DISEASES
Silicosis
A fibrogenic pneumoconiosis caused by inhaling crystalline free silica (quartz) dust; characterized by discrete nodular pulmonary fibrosis and, in more advanced stages, by conglomerate fibrosis and impaired respiratory function.
Etiology
Silicosis is the oldest known occupational lung disease. It usually follows long-term inhalation of small particles of free silica (silicon dioxide) in such industries as mining (lead, hard coal, copper, silver, gold), foundries, pottery making, and sandstone and granite cutting. Usually, 20 to 30 yr of exposure are necessary before the disease becomes apparent, though it develops in a much shorter time (< 10 yr) when the dust-dose is extremely high, as in such industries as tunneling, abrasive soap making, and sandblasting. The present standard for free silica in the industrial atmosphere is an 8-h tune-weighted average based on the percent of silica in the dust. The formula for calculating this threshold limit value (TLV) for respirable dust is: TLV = (10 mg per cu meter/% SiO2).
Pathology and Pathophysiology
Respirable particles of free silica (< 5 p. in diameter) are engulfed by alveolar macrophages. The macrophages ultimately die, hydrolytic enzymes are released, and fibrosis of the lung parenchyma occurs. The typical pathologic change initially is the formation of discrete hyahnized silicotic nodules distributed throughout the lungs. Later, coalescence of fibrosis results in conglomerate masses, contraction of the upper lung zones, and basilar emphysema with marked distortion of lung architecture. Ventilatory and gas exchange functions are affected, and ultimately there is reduced lung volume. This reduced lung volume distinguishes the overall physiologic pattern of silicosis from that of advanced chrome bronchitis or pulmonary emphysema. The striking pulmonary functional impairment occurs in association with progression to the severe late stages of silicosis. Respiratory failure is the ultimate consequence of silicosis, which may progress for some years even after exposure ceases.
When the dust-dose is extremely high and disease is accelerated, more uniform pathologic findings are found in me lung parenchyma. There is a diffuse interstitial reaction and, at times, filling of fine alveolar spaces with a protemaceous material similar to that found in alveolar proteinosis.
Symptoms, Signs, and Clinical Course
Patients with simple nodular silicosis have no respiratory symptoms and usually no respiratory impairment. They may cough and raise sputum, but these symptoms are due to industrial bronchitis and occur equally frequently m subjects who have normal x-rays. Though simple silicosis has little effect on pulmonary function, occasionally categories 2 and 3 lead to a slight reduction of lung volumes, but the values are seldom outside the predicted range. Conglomerate silicosis, in contrast, leads to severe shortness of breath, cough, and sputum. The seventy of the shortness of breath is related to the size of the conglomerate masses in the lungs; when they become extensive, the affected subject becomes completely disabled. As the masses encroaching on the vascular bed increase in size, pulmonary hypertension and right ventricular hypertrophy supervene. In advanced conglomerate silicosis, there may be signs of consolidation over the affected area and also of pulmonary hypertension. Eventually the person dies from cor pulmonale.
Pulmonary function abnormalities are frequent in complicated pneumoconiosis, especially in the later stages. These consist of decreased lung volumes and diffusing capacity, airways obstruction, and frequently pulmonary hypertension and desaturation. CO2 retention is unusual. Populations exposed to silica have 3 times the risk of the development of TB; generally, the more silica present in the lungs, the greater the risk. Silicotuberculosis resembles conglomerate silicosis radiologically, and the distinction can be made only by culturing the sputum. The sera of many silicotics contain lung autoantibodies and antmuclear factor.
Diagnosis
Silicosis is diagnosed from characteristic x-ray changes and a history of exposure to free silica. Simple silicosis is recognized by the presence of multiple, small, rounded or regular opacities in the chest film, and is subdivided into categories 1, 2, and 3 according to their profusion. Conglomerate silicosis is recognized by the development of an opacity > 1 cm in diameter on a background of category 2 or 3 simple silicosis. Numerous other diseases may resemble simple silicosis, including miliary tuberculosis, welder’s siderosis, hemosiderosis, and coal worker’s pneumoconiosis. The presence of eggshell calcification does, however, distinguish silicosis from most other occupational lung diseases (picture 8).
Picture 8. Their disease is thus recognized radiographically with multiple small nodules with an upper lobe predominance. Hilar adenopathy with “eggshell” calcification can be seen
Prophylaxis
Silicosis is preventable with effective dust control methods. Since dust suppression cannot reduce the risk in sandblastmg, extemal-air-supplied hoods should be used during sandblastmg. Such protection may not be available to personnel performing other jobs m the area (e.g., painting, welding). For this reason, substitute abrasive materials should ultimately replace sand m this operation. Medical surveillance of all exposed workers should include periodic chest x-rays.
Treatment
No effective treatment is known for silicosis. Persons with airways obstruction accompanying silicosis should be treated as if they had chronic airflow obstruction. Those who are exposed to sihca and who have a positive tuberculin test should be given isoniazid for at least 1 yr. Some authorities recommend lifetime treatment because the alveolar macrophage function is permanently compromised by silica.
coal worker’s pneumoconiosis (CWP)
(Coal Miner’s Pneumoconiosis; Black Lung Disease; Anthracosis)
Diffuse nodular deposition of dust in the lungs as a result of long-term exposure to bituminous or anthracite coal dust in coal mining.
Pathology and Pathophysiology
In simple CWP, coal dust is widely deposited throughout the lungs, leading to the development of “coal macules” around the respiratory bronchioles. Later on mild dilation also occurs m the same region. This dilation is known as focal emphysema; however, it does not extend to the alveoli and is not associated with airfiow obstruction. Because coal is relatively nonfibrogenic, distortion of lung architecture and functional impairment are minimal. However, a few miners with categories 2 or 3 simple CWP go on to develop progressive massive flbrosis (PMF). The latter is defined as development of a large opacity > 1 cm on a background of relatively advanced simple CWP. PMF may develop after exposure has ceased and may progress without further exposure, but not all subjects show progression. PMF encroaches on and destroys the vascular bed and airways, as does complicated silicosis, but this mass is amorphous and black. The development of PMF appears to be unrelated to the silica content of the coal; however, as in silicosis, antinuclear antibodies and lung autoantibodies may be present in the serum of the affected person.
Symptoms, Signs, and Diagnosis
CWP is not associated with any respiratory symptoms. Cough and sputum occur as frequently in men without x-ray evidence of this condition. When airways obstruction is present, then it is either due to coincident pulmonary emphysema from smoking, industrial bronchitis, or the presence of PMF. which is the only disabling form of CWP. A few minor abnormalities of the distribution of inspired gas are found in simple CWP, but these are not associated with respiratory symptoms. The diagnosis depends on a history of suitable exposure, usually at feast 10 yr underground, and the characteristic x-ray pattern of small rounded opacities m both lung fields (simple CWP) or, in PMF, a shadow > 1 cm in diameter occurring on a background of at least category 2 or 3 simple CWP (picture 9)
Picture 9. Progressive massive fibrosis. Chest imaging reveals small nodules with upper and posterior zone predominance. Hilar lymph node enlargement is not uncommon although eggshell calcification does not generally occur. When PMF occurs, these small nodules coalesce, forming opacities larger than 1 cm. These lesions are odd-shaped, usually bilateral, progressive, and may cavitate or become calcified
Prophylaxis and Treatment
CWP can be prevented by increasing the efficiency of dust suppression at the coal face. The development of PMF usually can be prevented by removing patients with x-ray changes typical of simple CWP from further coal dust exposure. There is no specific treatment; therapy is similar to that for nonspecific chronic obstructive disease.
ASBESTOSIS
A diffuse fibrous pneumoconiosis resulting from the inhalation of asbestos dust (fibrous mineral silicates of different chemical compositions).
Etiology Asbestosis is a consequence of long-term inhalation of asbestos fibers in the mining, mining, manufacturing, or application (e.g., of insulation) of asbestos products. The risk of developing asbestosis is related to the dose of asbestos dust to which the worker has been exposed. The incidence of lung cancer is also increased in asbestos-exposed workers. Although the asbestos exposure is usually less (asbestotic fibrosis is often absent), the risk is not entirely limited to cigarette smokers. Rare tumors of mesothelial lining surfaces (pleural or peritoneal mesotheliomas) have also been associated with asbestos exposure, though the exposure may have occurred many years earlier and may have been of very limited duration.
Pathology and Pathophysiology
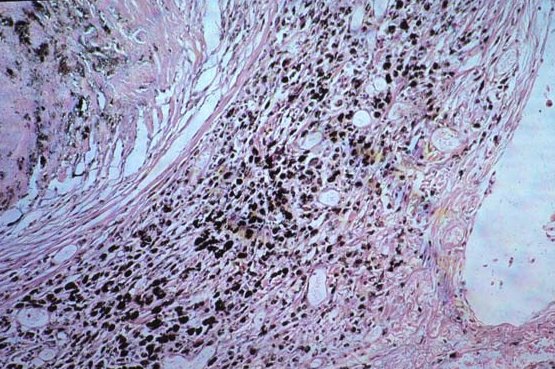
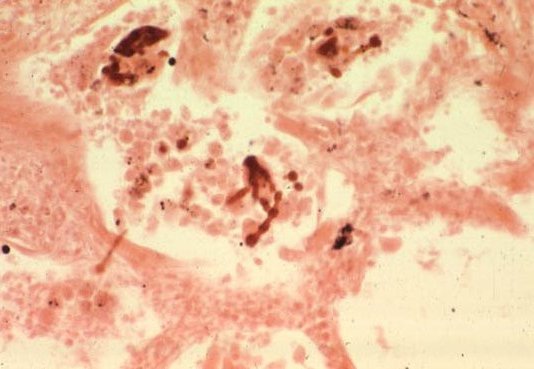
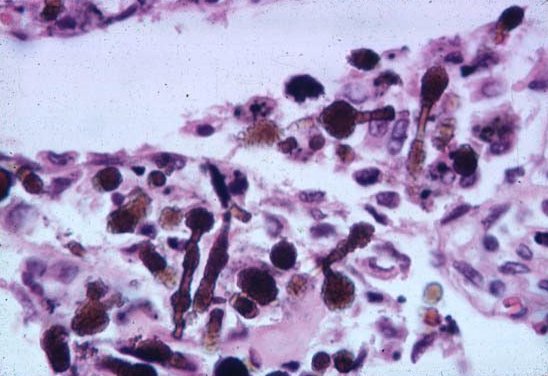
Asbestos fibers continually divide along their long axes, their diameter ultimately becoming very small (< 1 M). These small fibers can be inhaled deep into the lung parenchyma where they produce diffuse alveolar, interstitial, and pleura! fibrosis, resulting in reduced lung volumes and lung compliance (increased stiff-ness), and impaired gas transfer. Uncoated and coated (with an iron-protein complex) asbestos fibers (the latter are called “asbestos bodies”) may be present m lung tissue, with or without associated fibrosis. If there is no associated fibrosis, the presence of fibers in lung tissue indicates exposure only, not disease (picture 10).
Picture 10. The finding of asbestos or ferruginous bodies (above and below) in pathologic samples (arrows) help to support the diagnosis.
Symptoms and Signs
The patient characteristically notices the insidious onset of exertional dyspnea and reduced exercise tolerance. Symptoms of airways disease (cough, wheezing) are not usual but may occur m heavy smokers with associated chrome bronchitis. The chest x-ray reveals diffusely distributed irregular or linear small opacities, most prominent in the lower lung zones. Diffuse or local pleural thickening, with or without parenchymal disease, may also be visible. The process is progressive and symptoms become more severe in association with advancing x-ray and physiologic abnormalities. Ultimately, respiratory failure with marked impairment in oxygenation occurs.
Mesothelial tumors associated with asbestos exposure are invariably fatal. Bloody effusion with pain is often present. Spread is usually by local extension or, rarely, distant metastases.
Diagnosis
The diagnosis of asbestosis requires a history of occupational exposure, and x-ray (picture 11), clinical, and physiologic evidence of diffuse pulmonary fibrosis. Histologic confirmation is rarely necessary or indicated. Mesothelioma is more difficult to diagnose and may be confirmed only by biopsy or autopsy While the diagnosis of bronchogenic carcinoma can be made readily, the association with asbestos exposure in any individual case presents formidable medical and legal problems.
Prophylaxis and Treatment
Asbestosis is preventable, primarily by effective dust suppression in the work environment Marked reduction in asbestos exposure has reduced the incidence of asbestosis, and further industrial hygiene advances are likely essentially to eliminate this disease. Once dust is controlled to a point where asbestosis is no longer a problem, evidence suggests that the excess risk of lung cancer m asbestos workers will decrease. The most effective preventive measure can be taken by the worker himself, however, by abstaining from cigarette smoking. Since the dose of asbestos exposure necessary for the development of mesothelioma is much less, the prevention of this tumor cannot be confidently predicted.
No specific therapy is available for asbestosis. Treatment is symptomatic. Surgical resection occasionally produces cure for bronchogenic carcinoma, but is ineffective in treating mesothelioma.
Picture 11. Computed tomography of the chest demonstrating pleural plaques due to asbestos exposure
beryluosis
(Beryllium Disease, Poisoning, or Granulomatosis)
A generalized granulomatous disease with pulmonary manifestations, caused by inhalation of dust or fumes containing beryllium compounds and products.
Etiology
Beryllium exposure used to be common in a wide variety of industries including beryllium mining and extracting, electronics, chemical plants, and the manufacture of fluorescent light bulbs. However, demand has dropped and beryllium finds its main use now in the aerospace industry. Berylhosis differs from most of the other pneumoconioses in that it appears to be a hypersensitivity disease and occurs only in a small proportion (about 2%) of those exposed. Exposure may be relatively brief, and the onset of the disease may be delayed as much as 10 or 20 yr. A few persons with berylliosis have been reported among those living in the vicinity of beryllium refineries.
Pathology and Pathophyslology
Acute berylliosis resembles a chemical pneumonitis, but other organs such as the skin and conjunctiva may be involved. Pathologic changes in the lung include nonspecific infra-alveolar edema and diffuse parenchymal inflammatory infiltrates. Early granuloma formation with mononuclear and giant cells may also occur. The hallmark of chronic berylliosis is a diffuse pulmonary and hilar lymph node granulomatous reaction histologically indistinguishable from sarcoidosis.
Symptoms, Signs, and Diagnosis
Patients with acute beryllium disease often have dyspnea, cough, weight loss, and a highly variable chest x-ray pattern, usually indicating diffuse alveolar consolidation. In chronic disease, patients complain of insidious and progressive exertional dyspnea. The chest x-ray shows a pattern of diffuse infiltrations, often with hilar adenopathy, resembling the pattern seen in sarcoidosis. Diagnosis depends on a history of exposure and the appropriate clinical manifestations. However, in most instances it is impossible to distinguish berylliosis from sarcoidosis in the absence of sophisticated immunologic technics.
Prognosis
Prognosis in the acute disease is good, but the chronic form often results in progressive loss of respiratory function. Right heart strain may result, with death from cor pulmonale.
Prophylaxis
The substitution of harmless compounds in the manufacture of fluorescent lamps has prevented many cases of the disease. Some exposure to fluorescent tubes manufactured prior to 1949 still occurs, often from tubes broken during salvage operations. Industrial dust suppression is the basis of prevention in other types of beryllium exposure, but its efficiency is imperfect. The disease (both acute and chronic) must be promptly recognized and affected workers removed from further beryllium exposure.
Treatment
Treatment of acute disease is generally symptomatic. The clinical manifestations are usually short-lived and completely reversible. Chronic beryllium disease may respond to corticosteroid therapy (e.g., full doses of prednisone 60 mg/day orally, gradually reducing the dose after 2 to 3 wk to maintenance levels of 10 to 15 mg/day, with complete tapering of medication once maximal improvement has resulted). Corticosteroids have been especially effective in relatively early chronic cases, resulting in remissions apparent not only clinically, but also physiologically and radiologically. Such improvement has been maintained for many years.
SARCOIDOSIS
A multisystem granulomatous disorder of unknown etiology, characterized histotogically by epithelioid tubercles involving various organs or tissues, with symptoms dependent on the site and degree of involvement.
Ethiology and incidence
The cause is unknown. A single provoking agent (e-g., a slow virus) or disordered defense reactions triggered by a variety of insults may be responsible; genetic factors may be important. Sarcoidosis occurs predominantly between ages 20 and 40 and is most common among northern Europeans and American blacks. The incidence in some advanced countries exceeds that of TB.
Pathology
The characteristic histopathologic findings are multiple noncaseating epithelioid granulomas, with little or no necrosis, that may resolve completely or proceed to fibrosis. They occur commonly in mediastinal and peripheral lymph nodes, lungs, liver, eyes, and skin, and less often in the spleen, bones, joints, skeletal muscle, heart, and CNS.
Symptoms and signs
Symptoms depend on the site of involvement and may be absent, slight, or severe. Function may be impaired by the active granulomatous disease or by secondary fibrosis. Fever, weight loss, and arthralgias may be initial manifestations. Persistent fever is especially common with hepatic involvement. Peripheral lymphadenopathy is common and usually asymptomatic. Even insignificant nodes may contain characteristic tubercles.
Skin lesions (plaques, papules, and subcutaneous nodules) frequently are pesent in patients with severe chronic sarcoidosis. Nasal and conjunctival mucosa granulomas may occur (picture 12-13). Erythema nodosum with fever and arthralgias is a frequent manifestation in Europe, but less common in the USA.
Picture 12. Nails injured by Sarcoidosis
Picture 13. Skin lesion in Sarcoidosis patient
Mediastinal adenopathy often is discovered by routine chest x-ray. X-ray findings of bilateral hilar and right paratracheal adenopathy are virtually pathognomonic; adenopathy occasionally is unilateral. Diffuse pulmonary infiltration may accompany or follow the adenopathy; this infiltration may have a diffuse find ground-glass appearance on x-ray, may occur as reticular or miliary lesions, may be present as confluent infiltrations or large nodules that resemble metastatic tumors. Pulmonary involvement, which may also occur without visible adenopathy, is usually accompanied by cough and dyspnea, but these symptoms may be minimal or absent. Pulmonary fibrosis, cystic changes, and cor pulmonale are results of longstanding progressive disease (picture 14-16).
Picture 14. Twenty-eight year-old female with a history of minimal shortness of breath. The most common parenchymal pattern of sarcoidosis is a reticulonodular configuration, correlating with the non-caseating granulomas noted at pathology. Slightly less common is alveolar pattern made up of diffuse, small, indistinct opacities due to the alveolar filling with mononuclear cells.
Picture 15. CHEST X-RAY, SARCOIDOSIS – There is nodular hilar lymphadenopathy represented by hilar widening.


Picture 16. Enlaged lymphonodes due to sarcoidosis
Hepatic granulomas are found in 70% of patients examined by percutaneos biopsy, even if patients are asymptomatic with normal liver function tests. Hepatomegaly is noted in fewer than 20% of patients; progressive and severe hepatic dysfunction with portal hypertension and esophageal varices is rare.
Granulomatous uveitis occurs in 15% of cases; it is usually bilateral, and may cause severe loss of vision from secondary glaucoma if untreated. Retinal panpblebitis, lacrimal gland enlargement, conjunctival infiltrations, and keratitis sicca occasionally are present. Myocardial involvement may cause angina, congestive failure, or fatal conduction abnormalities. Acute polyarthritis may be prominent; chronic periarticular swelling and tenderness may be associated with osseous changes in the phalanges. CNS involvement is of almost any type, but cranial nerve palsies (especially facial paralysis) are most common. Diabetes insipidus may occur. Hypercalcemia and hypercalcfuria may cause renal calculi or nephrocalcmosis with consequent renal failure, but prednisone therapy has reduced the frequency and importance of disordered calcium metabolism.
Laboratory Findings
Leukopenia frequently is present. Hyperglobulinemia is common among blacks. Elevated serum uric acid is not uncommon, but gout is rare. Serum alkaline phosphatase may be elevated as a result of hepatic involvement. Depression of delayed hypersensitivity is characteristic, but a negative second-strength tuberculin reaction reliably excludes a complicating TB.
Pulmonary function tests show restriction, decreased compliance, and impaired diffusing capacity. CO2 retention is uncommon, since ventilation rarely is obstructed except in patients with endobronchial disease or in late stages with severe pulmonary fibrosis. Serial measurements of pulmonary function are a guide to treatment and to the course of the disease.
Diagnosis
A clinical diagnosis may be made in asymptomatic patients with typical chest x-ray findings, but the diagnosis must be considered in the presence of the symptoms and signs described above even if (as in about 10% of patients) the chest x-ray is normal. Tissue biopsy, with microbiologic as well as histologic examination, is essential if symptoms are present and corticosteroid therapy seems indicated. When superficial or palpable lesions (e.g., in skin. lymph nodes, palpebral conjunctiva) are present, biopsy is positive in 87% of specimens.
When physical examination is negative, transbronchial biopsy by fiberoptic bronchoscope is the best initial procedure for securing histologic evidence of sarcoidosis. This technic has shown granulomas in 60 to 90% of patients, whether the chest x-ray reveals pulmonary infiltration or hilar adenopathy alone.
If this approach is not available or fails to show granulomas, other possible biopsy sites include me mediastinum, which can be approached by mediastinotomy or mediastinoscopy; the lungs, approached by intercostal biopsy; or random biopsies of skeletal muscle and conjunctiva. Liver biopsy shows granulomas in 70% of cases, and can be useful. Scalene fat-pad biopsy is obsolete in view of the higher yields of other methods.
Local sarcoid reactions in a single organ and granulomas due to infection or hypersensitivity must be excluded. In questionable cases, histologic evidence of 8ranulomas should be sought in more than one site. The Kveim reaction, a granulomatous reaction appearing 4 wk after intradermal injection of extracts of spleen or lymph node, is positive in 50 to 60% of patients, but reliable antigens are not available in the USA.
Anigiotensin converting enzyme (ACE) is elevated significantly in sera of patients with sarcoidosis, presumably reflecting macrophage activity. Tissue levels are highest in sarcoid lymph nodes rather than in pulmonary tissues. Elevations greater than 2 standard deviations occur in 60% of patients with sarcoidosis, but these elevations are also seen in 10% of patients with TB or lymphoma; therefore elevated ACE has limited diagnostic value, but may prove useful in following the course of sarcoidosis.
TB still must be distinguished from sarcoidosis, but aspergillosis and cryptococcosis are now more frequent complications of sarcoidosis. Hodgkin’s disease also must be excluded. It is uncertain whether the typical sarcoid granulomas found in 5% of liver biopsies done for staging of Hodgkin’s disease indicate 2 concurrent diseases or a sarcoid reaction to the neoplasm.
Course and Prognosis
Evaluating treatment is difficult, since spontaneous improvement or clearing is common. Massive hilar adenopathy and extensive infiltrates may disappear in a few months or years. Mediastinal adenopathy persists without change for many years in about 10% of cases. In 1/3 of the patients, complete clearing of the disease occurs; another 1/3 recover, but with minor residua; in the remaining 1/3, progressive disease requires treatment. Mortality is < 5%. Gradual pulmonary fibrosis, leading to pulmonary insufficiency, pulmonary hypertension, and cor pulmonale, is the leading cause of disability and death; pulmonary hemorrhage from aspergillosis is the second most common cause of death.
Treatment
No available therapeutic agents have been shown to prevent progressive tissue damage and fibrosis of the lungs. Corticosteroids accelerate clearance of symptoms, physiologic disturbances, and roentgenographic changes; but after 5 yr no difference is demonstrable between treated and untreated patients. Asymptomatic hilar or peripheral adenopathy needs no treatment. Corticosteroid therapy should be given to suppress troublesome or disabling symptoms such as dyspnea, severe arthralgia, or fever, and should be started promptly if active ocular disease, respiratory failure, hepatic insufficiency, cardiac arrhythmia, CNS involvement, or hypercalcemia is present. Prednisone therapy is required by 1/3 of white patients and 2/3 of black patients with sarcoidosis.
Prednisone 40 to 60 mg/day orally may be given when a prompt effect is desired, but doses of 10 to 15 nag/day by mouth usually are adequate to control the inflammatory reaction. If doses > 15 mg/day are given, alternate-day schedules should be employed. Treatment may be needed for weeks, for years, or indefinitely. Maintenance doses of 5 to 10 mg/day are surprisingly effective in controlling symptoms and radiologic changes in many chronic cases. Clinical examination, x-rays, and pulmonary function studies should be made at frequent intervals when dosage is being reduced or medication terminated. Serious complications of corticosteroid therapy are infrequent with low-dose therapy in this disease. Concomitant isoniazid therapy, 300 mg/day for a year, is indicated only for the few patients given corticosteroids who have positive tuberculin skin tests.
Methotrexate and chlorambutil occasionally are effective in sarcoidosis, but dramatic improvement with these agents is rare. They deserve a trial only when corticosteroids fail or are contraindicated.
IDIOPATHlC INFILTRATIVE DISEASES OF THE LUNGS
A spectrum of disorders with different etiologies but similar clinical features and diffuse pathologic changes that affect primarily interalveolar interstitial tissue. Interstitial infiltration is characterized in its acute phase by abnormal accumulation of histiocytes, lymphocytes, plasma cells, eosinophils, and exudate in alveoli and bronchioles. Hyperplasia of bronchiolar or alveolar epithelium may be present at a later stage. If the disorder progresses, the exudate may become organized, and necrosis, scarring, and reepithelialization of alveolar septae may take place. The whole process may ultimately lead to extensive fibrosis, progressive destruction of lung and formation of cysts (“honeycombing”).
Idiopathic pulmonary fibrosis (usual interstitial pneumonia [UIP], diffuse fibrosing alveolitis, Hamman-Rich syndrome): when the etiology leading to pulmonary fibrosis cannot be defined (about 50% of cases), the term “idiopathic” is used.
Desquamative interstitial pneumonia (DIP) resembles idiopathic pulmonary fibrosis, but the histology tends to be more uniform: the cellular infiltrate is more sparse and less pleomorphic. There is striking hyperplasia of type II pneumocytes and filling of air spaces with macrophages. It has been argued that the separation of DIP from idiopathic pulmonary fibrosis is artificial because both histologic patterns can be found frequently in the same lung (probably representing different phases of the same process). However, the clinical recognition of DIP is important because the process is associated with a better prognosis and a better response to systemic corticosteroids.
Symptoms and Signs
Symptoms and signs vary with the extent of pulmonary infiltration, its rate of progress, and with the presence of complications such as pulmonary infections or cor pulmonale. Pulmonary symptoms may be few, but exertional dyspnea of insidious onset is almost invariably present. Cough is usually not prominent, but it is more likely to be present when there is secondary bronchial infection. Anorexia, weight loss, fatigue, weakness, and vague chest pains are common. Physical signs may be absent early in the course, but, as the disease progresses, tachypnea and labored breathing are observed and chest examination reveals prominent breath sounds and end-inspiratory crackles at lung bases. With progression, cyanosis, cor pulmonale, and clubbing may appear.
Laboratory Findings
Routine laboratory studies are not helpful. Polycythemia may be present secondary to chronic hypoxemia. Chest x-rays may be normal even in the presence of significant symptoms or functional abnormalities. X-ray changes tend to be more prominent at the bases and may include diffuse or patchy “ground-glass” haziness, linear markings, rounded opacities, small cystic lesions (honeycombing), evidence of reduced lung volumes, and signs of pulmonary hypertension. Pulmonary function studies reveal a restrictive ventilatory defect with reductions in both vital capacity and residual volume. The coefficient of retraction (maximum static transpulmonary pressure/total lung capacity) can be increased. Arterial blood gases show a low Paco2 denoting hyperventilation at rest and a decrease in Pao2. The abnormal increase in PaCO2 at rest may be exacerbated during exercise. The diffusing capacity for CO is usually reduced. These functional abnormalities can worsen as the disease progresses.
Diagnosis
Diagnosis is made by recognizing the clinical features, demonstrating the presence of a diffuse interstitial disorder, and excluding a specific etiology. Because the amount of tissue obtained by transbronchial biopsy is frequently insufficient, open lung biopsy is recommended for the identification of DIP. Open pulmonary biopsy is not indicated when there is radiographic honeycombing.
Prognosis
The outcome varies with the etiology and the rate of progression. Some patients may die within a month, while others survive many years. The mortality is smaller and the mean survival greater when histologic features of DIP are present on open lung biopsy.
Treatment
A trial of systemic corticosteroids is indicated in patients without evidence of extensive fibrosis. Prednisone 40 to 60 mg/day usually is given with gradual reduction of the dose to maintenance levels (10 to 30 mg every other day). The response to therapy is followed by serial chest x-rays and appropriate lung function tests. Gallium citrate Ga 67 lung scanning and serial analysis of cellular content of bronchoalveolar lavage fluid may prove to be useful to detect and follow the response of the inflammatory process to therapy. A few patients who have not improved on prednisone have shown improvement with azathioprine 3 mg/kg/day, but experience with this agent is limited. Other treatment is supportive and palliative. 02 in high concentrations may help combat hypoxemia. Antibiotics are required if secondary bacterial infection occurs. Digitalis and diuretics are used to treat heart failure.
Differential diagnostics of lung injuries in connective tissue disease
In connective tissue disease symptoms may include: shortness of breath, especially with exertion, fatigue and weakness, loss of appetite, loss of weight, dry cough, that does not produce phlegm, discomfort in chest, labored breathing, hemorrhage in lungs.
Examinations of lungs reveals:
Pleuropulmonary manifestations are common. The incidence varies from 20-85%.
Pleuropulmonary complications include pleural effusion, interstitial pulmonary fibrosis, pulmonary arterial hypertension, pulmonary vasculitis, pulmonary thromboembolic phenomena, aspiration pneumonia, serositis, and hypoventilatory failure.
Pulmonary injuries are characterized by the presence of ground-glass attenuation, nonseptal linear opacities, and peripheral and lower lobe predominance.
- Ill-defined centrilobular opacities may be evident.
Chest radiography may depict pleural thickening, fibrosis, and pericarditis
- Pulmonary function tests may reveal ventilation disorders of the restrictive type.
- These include a diminished vital capacity, a normal residual volume, and a diminished total lung capacity.
- Respiratory function abnormalities usually are coexistent with the radiographic findings
The main role in differential diagnostics based on clinic of multiorgan involvment, immunological and histological findings, that are very specific in case of connective tissue diseases.

