Theme: Demyelination diseases of nervous system: multiply sclerosis (MS), acute
disseminated encephalomyelitis (ADEM), optic encephalomyelitis,
encephalomyelopolyradiculoneuritis, polyencephalomyelitis, disseminated myelitis. Acute myelitis.
Multiple
sclerosis is a demyelinating disease of the central nervous system (CNS) caused
by an autoimmune reaction that is the result of a complex interaction of
genetic and environmental factors.
Epidemiology
Multiple
sclerosis is more common in women, with a female:male ratio of 2.1:1. The
disease is rare in children and has a peak incidence at 30 to 33 years of age,
with a decline in the late forties. The upper limit of age at onset has usually
been accepted as 59 years, but late onset of multiple sclerosis after the age
of 60 years is reported. However, these cases probably represent late
occurrence of overt symptoms in individuals who have had the disease in a subclinical or unrecognized fashion for many
years.
The natural
history of multiple sclerosis has been studied extensively and it is clearly a
disease of temperate zones, with an increase in the prevalence gradient south
to north in the northern hemisphere, and north to south in the southern
hemisphere. Three zones of high, medium, and low prevalence rates can be recognized.
High-frequency prevalence rates of more than 30 per 100,000 population occur in
areas lying between latitudes 45° and 65° north or south. This includes
northern Europe, southern
These figures
cannot be attributed to climate alone, however, because there are
well-documented ethnic differences. Multiple sclerosis predominantly affects
people of northern European extraction. In the
Such
findings suggest a genetic factor in multiple sclerosis, and family studies
have demonstrated that the risk of multiple sclerosis is increased for relatives
of patients with the disease. Studies in
At the
present time, evidence suggests that multiple sclerosis is influenced by
several genes, the major histocompatibility (MHC)
complex class 2, HLA-DRII allele having
the strongest association with the disease in northern European populations.
However, the association of different class 1 and class 2 alleles has been reported in other populations.
Consequently, the current impression is that multiple sclerosis is probably polygenic, the result of complex genetic
factors involving the interaction of genes and coding within and outside of the
MHC complex. However, epidemiologic studies
in
At the
present time, it is considered probable that both genetic and environmental
factors are involved in multiple sclerosis, with infection as the major
environmental agent, in that both viral and bacterial infections can initiate
or precipitate attacks of multiple sclerosis. Evidence for a direct involvement
of a viral agent such as human T-cell lymphotropic virus (HTLV)-l, herpes
simplex virus (HSV)-l, HSV-6, scrapie,
parainfluenza virus 1, measles virus, coronavirus, simian virus,
chimpanzee cytomegalovirus, and LM7 retrovirus in multiple sclerosis is less
compelling. The role of environmental factors other than infection has been
studied, including the relationship of trauma and multiple sclerosis,
indicating that patients are at no greater risk to experience an exacerbation
of multiple sclerosis after trauma than at other times, nor is it likely that
trauma is ever a causal factor in initiating the disease process. The role of
emotional stress in multiple sclerosis is more controversial because stress is
difficult to define and quantitate.

Etiology,
Pathology, and Pathogenesis
The etiology
is unknown. The pathological changes in multiple sclerosis show variation,
depending on the age of focal demyelination (the plaque). In the acute state,
there is active demyelination with accumulation of sudanophilic myelin breakdown products. The area is edematous and there is marked perivascular cuffing around veins and venules by lymphocytes and macrophages. Plaques are frequently located in
the periventricular distribution,
particularly in the cerebral hemispheres, but plaques can occur at any site in
the white matter and often penetrate into the gray matter of the cortex and
deeper gray matter structures in the cerebrum and cerebellum. Because multiple
sclerosis is a disease of the CNS, plaque formation is not uncommon in the brainstem, cerebellum, spinal cord, and the
optic nerves, which are structurally part of the CNS, in which the oligodendrocytes are anti-genically similar to
the oligodendrocytes in the spinal cord.
All multiple
sclerosis lesions show a variable degree of axonal
loss, ranging from 10 to 20 percent in milder forms of the disease, to
80 percent in severe, acute multiple sclerosis.
Epidemiologic studies support the concept
that multiple sclerosis results from an aberrant immune reactivity occurring in
a genetically susceptible host who has acquired a specific, or one or more
nonspecific, neurotropic infections at a
critical age. Genetic susceptibility is thought to be associated with genes
within or close to the HLA-DR DQ subregion, located in the short arm of
chromosome 6. This primary infection results in a self-sustaining,
organ-specific autoimmune disorder that remains latent until activated by a subsequent
infection years after the primary event. An alternative explanation suggests
that the mechanism is one of persistent systemic viral infection, which
contributes to the changes in the CNS periodically, or to a persistent CNS
viral infection that is targeted by T cells unpredictably, resulting in an
inflammatory response producing myelin damage
(bystander response). The common factor in any one of these theoretical
situations is the activation of an autoimmune event within the CNS, directed
against myelin antigen-specific T cells,
but no specific antigen has as yet been identified. Antibodies to myelin basic protein appear to be the most
frequent finding in multiple sclerosis, but reactivity to other myelin antigens is a possibility. Candidates
include proteolipid protein, the most abundant myelin
protein in humans, myelin oligodendrocyte
glycoprotein, myelin-associated protein, minor myelin proteins, or heat shock proteins.
The
autoimmune reaction probably begins with a systemic infection that is associated
with liberation of γ-interferon, resulting in activation of CD4 T lymphocytes.
The lymphocytes attach to adhesion molecules on the surface endothelium of postcapillary
venules, roll along the surface of the endothelium,
producing endothelial cell
activation, followed by passage of the CD4 T lymphocytes into the CNS—in
effect, disrupting the blood-brain barrier. Once within the CNS, the
T-lymphocyte receptors respond to antigen presented by MHC class 2 molecules on
macrophages and astrocytes, but oligodendrocytes are
usually preserved at this stage. The antigen T-cell receptor interaction is
followed by stimulation of helper T cells, T-cell proliferation, and B-cell and
macrophage activation, with release of cytokines such as •γ-interferon, tissue
necrosis factor, interleukin-12, and proteases. The cytokines induce a local inflammatory reaction with further
disruption of the blood-brain barrier, followed by a major influx of CD4
lymphocytes and monocytes into the
lesion. Myelin damage results
from the combined effect of cytotoxic cytokines,
particularly tissue necrosis factor and cytotoxic
cells.
Oligodendrocytes appear to
survive and proliferate in the presence of acute demyelination and interact
with hypertrophied astrocytes, an
association that may represent a short-term protective mechanism.
Consequently, active demyelination and remyelination can occur in acute
lesions. Oligodendrocyte depletion in
chronic multiple sclerosis lesions may be a slow, insidious process, spanning a
protracted period during which there is gradual cell dropout.
Classification of Multiple
Sclerosis
Although
multiple sclerosis can affect any site in the CNS, it is possible to recognize
eight types of the disease (Table 7-1):
1. Relapsing-remitting multiple sclerosis.
This is the classical form of multiple sclerosis that often begins in the late
teens or twenties with a severe attack followed by complete or incomplete
recovery. Approximately 70 percent of patients with multiple sclerosis
experience a relapsing-remitting course initially.38 Further
attacks occur at unpredictable intervals, each followed by increasing
disability. The relapsing-remitting pattern tends to change into the secondary
progressive form of the disease in the late thirties.
2. Primary progressive multiple sclerosis. The
disease runs a steady deteriorating course that may be interrupted by periods
of quiescence without improvement. The rate of progression is variable; at its
most severe, this form of multiple sclerosis can terminate in death within a
few years. In contrast, the more chronic form of progressive multiple sclerosis
is similar to the benign form of the disease.
Table 7-1
Eight Types
of Multiple Sclerosis
1. Relapsing-remitting
2. Primary progressive
3. Secondary progressive
4. Relapsing progressive
5. Benign
6. Spinal form
7. Neuromyelitis
optica (Devic disease)
8. Marburg variant
3. Secondary progressive multiple sclerosis.
The relapsing-remitting form of the disease frequently develops into secondary
progressive multiple sclerosis after a variable period of time but usually in
the late thirties.
4. Relapsing progressive multiple sclerosis.
Occasional cases are encountered where patients with a progressive form of
multiple sclerosis have superimposed relapses with no significant recovery.
5. Benign multiple sclerosis. About 20 percent
of cases have the benign form of multiple sclerosis. This may be defined as
multiple sclerosis in which the patient is able to function at the level of
full employment or provide care of home and family independently 10 years after
the appearance of the first symptoms. It is extremely unlikely that these patients
will ever be incapacitated by the disease and they should continue to live a
full life span with only occasional minor symptoms.
The
existence of a benign form of multiple sclerosis increases the importance of
recording the date of the first symptoms in patients who appear to have few
residual abnormal signs several years after the onset of the disease. These patients
may be informed that they have a benign form of multiple sclerosis 10 years
following their first recorded symptom and that the benign course will continue
in the years ahead.
6. Spinal form of multiple sclerosis. This form of multiple sclerosis presents with
symptoms and signs of predominantly spinal cord involvement from the beginning
and maintains this pattern. There may be a clear-cut pattern of relapse and
remission initially, followed by the secondary progressive form of the disease
after several years, or the presentation may be one of steady deterioration
from the onset.
7. Neuromyelitis optica (Devic syndrome). Most cases of
this syndrome are believed to be examples of multiple sclerosis presenting
with acute transverse myelitis followed by optic neuritis. Many patients
follow a relapsing-remitting course indistinguishable from multiple sclerosis.
8. Marburg variant. This rare and malignant
form of multiple sclerosis is associated with a fulminating course of
progressive impairment of consciousness, severe visual loss, dysarthria, dysphagia, respiratory
insufficiency, and rapid deterioration. It is indistinguishable from acute
disseminated encephalomyelitis. The
Marburg variant may result from the autoimmune process of multiple sclerosis
occurring in an individual
with developmentally immature myelin
basic protein.
Clinical
Features
The
diagnosis of multiple sclerosis is based on the clinical demonstration of
multiple levels of involvement of the CNS. Symptoms may be grouped under
several headings.
Sensory
Symptoms
Sensory
symptoms are the most common symptoms experienced by patients with multiple
sclerosis. These symptoms are often forgotten or ignored by both patient and
physician. Even prolonged sensory symptoms fail to evoke concern, in contrast
to the almost immediate response that occurs with weakness or paralysis.
Consequently, many patients date the onset of multiple sclerosis from the first
appearance of weakness, visual loss, or other symptoms of dramatic onset rather
than forgotten or poorly recorded sensory symptoms.
Sensory
symptoms include impairment of sensation (hypesthesia), tingling (paresthesias), and uncomfortable sensations (dysesthesias) often referred to as
"burning," which may be present for days, weeks, or months without
objective abnormalities. All patients with suspected multiple sclerosis should
be carefully questioned about the occurrence of previous sensory symptoms.
Motor
Symptoms
Paralysis or
paresis of upper or lower limbs is the most common presenting symptom in
patients with multiple sclerosis. Paraparesis is
a common early complaint when the patient gives a history of increasing
weakness and stiffness of the lower extremities, associated with progressive
impairment of gait. Examination shows signs of upper motor neuron involvement
with spasticity, increased tendon
reflexes, and extensor plantar responses. These findings may be quite subtle in
the early states of the disease.
Visual Symptoms
Optic
neuritis presents with sudden visual loss and pain on eye movement and a
unilateral headache. The condition may be followed by a rapid progression to
total loss of vision in the affected eye (Fig. 7-1) When vision is preserved,
there is monocular blurring; a central, paracentral,
or centrocecal scotoma; and
impairment of color vision. In optic neuritis, there may be edema of the optic
discs, but the appearance can be normal in retrobulbar
neuritis, when the inflammatory response is localized and located
proximally in the optic nerve. Temporal pallor is a later development because
of demyelination of the maculopapular bundle. However, subclinical involvement of the optic nerve can occur and may be
present without symptoms, when the patient first presents with signs of
multiple sclerosis or may be identified when the patient presents with the
first symptoms of optic neuritis, indicating prior subclinical involvement of the optic nerve.
Recovery
from optic neuritis is unpredictable. Many patients experience no further
problems for several years and then develop symptoms of brain or spinal cord
involvement, indicating multiple sclerosis. However, not all optic neuritis is
multiple sclerosis, and only 50 percent of adolescents and young adults who
present with the sudden onset of optic neuritis subsequently develop
multiple sclerosis.

Those with a
large time interval between the optic neuritis and the development of
additional symptoms have a better prognosis. The clinician should always
inquire about the possibility of preceding symptoms in any patient who presents
with optic neuritis because the presence of symptoms some years before the
more dramatic visual symptoms might indicate a more benign prognosis.
Diplopia is indicative of third or
sixth nerve involvement in the brainstem. The
fourth nerve is rarely involved in isolation. Intemuclear ophthalmoplegia is pathognomonic of multiple sclerosis and rarely has another
etiology. Unilateral or bilateral Marcus Gunn pupil is often present in optic
or retrobulbar neuritis.
Bladder
Involvement
The early
symptoms of bladder dysfunction consist of occasional urgency of micturition
followed by mild nocturia occurring once
a night. The events gradually increase in number, disturbing the sleep of the
patient and the bed partner. There is a concomitant increase in urinary
frequency and urgency during the day, ultimately resulting in incontinence.
Impaired bladder control is usually the result of spinal cord involvement in
multiple sclerosis and decreasing bladder control usually parallels increasing paraparesis. However, this is not an inevitable
relationship and some patients retain adequate bladder function even when paraplegic.
The
anatomical center for bladder control lies in the tegmentum of the pons. The
center is under the influence of a higher level of control located in the
medial aspect of the frontal lobes. Thus, the frontal lobes can signal the pontine bladder control center to inhibit
bladder function or to initiate bladder emptying at will or when convenient.
The pontine center then inhibits or
permits bladder contraction through connections that traverse the spinal cord
and exit through the parasympathetic outflow
in the S2-S4 sacral nerves supplying the bladder.
A number of
abnormal responses are associated with interruption of bulbar or spinal cord connections in multiple sclerosis.
1. Detrusor hyperreflexia.
The interruption of the afferent impulses from the detrusor muscle of the bladder to the pontine micturition center by spinal cord disease results in an
uninhibited reflex at the sacral cord level. The detrusor muscle is no longer inhibited as bladder volume increases
and detrusor contraction is initiated in
response to smaller volumes of urine, resulting in increasing frequency.
2. Detrusor sphincter dyssynergia. The normal
pattern of voiding is disturbed. The normal relaxation of the external
sphincter is impaired and detrusor contraction
is poorly coordinated and accompanied by contraction rather than relaxation of
the external sphincter. The result is poor flow of urine and incomplete
emptying of the bladder.
3. Detrusor hypocontractility is a failure to
empty the bladder secondary to insufficient detrusor
pressure or a fading contraction on voiding.
Cerebellar Symptoms
Tremor, dysarthria, truncal ataxia, and limb ataxia are frequent symptoms of multiple
sclerosis. Occasionally, cerebellar dysfunction
is the dominant feature in multiple sclerosis, when a patient presents with
adequate vision and muscle strength shows serious disability from the cumulative
effects of the several forms of cerebellar
ataxia.
Brainstem Symptoms
Many
patients with multiple sclerosis develop signs of brainstem involvement. Involvement of the oculomotor and sixth cranial
nerves as they traverse the substance of the brainstem
results in diplopia. Intemuclear ophthalmoplegia due to involvement of the
interaxial fibers connecting the third, fourth, and sixth nerve nuclei is not
uncommon. Sensory loss over the face indicates involvement of the afferent
fibers entering the pons from the trigeminal nucleus. Facial weakness may be due
to involvement of the seventh nerve in the pons.
Episodic dysarthria and dysphagia indicate involvement of the vagus
nerve in the medulla, and dysarthria may
be due to involvement of the vagus nerve, the glossopharyngeal
nerve, and the hypoglossal nerve
as they course through the medulla. Involvement of the corticospinal tracts in the brainstem
can produce a progressive spastic quadriparesis; involvement of the cerebellar connections results in limb and truncal ataxia.
Spinal Cord
Symptoms
Most
patients with established multiple sclerosis have signs of spinal cord
involvement. These signs include some degree of spastic paraparesis with increased tone in both lower limbs, bilateral
ankle clonus, increased tendon reflexes, and bilateral extensor plantar
responses. It is not unusual to see a progression of paraparesis with increasing disability. This does not necessarily
indicate progression of the disease but may be due to progressive gliosis of plaques in the spinal cord. This
scarring produces increasing traction on and destruction of axons descending from higher centers in the CNS
and results in increasing spasticity and paraparesis.
Abnormal
Bowel Function
Constipation
may be a major problem in advanced multiple sclerosis. Bowel incontinence can
be a devastating experience to a patient with multiple sclerosis, particularly
if the loss of control occurs in a social situation or in a crowded area such
as a shopping center. Many patients react to the incident with reluctance to
leave home and are extremely apprehensive if they do so.
Memory
Deficits and Dementia
Impaired cognitive
processing is not unusual in multiple sclerosis when patients often display a
verbal working memory deficit owing to a central processing problem. This has a
significant impact on reading or other tasks that require the maintenance of
verbal information over a short period of time. Dementia occurs in approximately
50 percent of cases, with less cognitive impairment in patients with
relapsing-remitting disease than in those with the progressive form. The
disease can, however, remain predominantly spinobulbar in form, with little
involvement of the white matter in the cerebral hemispheres and preservation of
intellect. Patients with demyelination in the periventricular
white matter of the brain often show an explosive emotional response
with inappropriate laughter or occasional crying during conversation. This
condition results from the interruption of an inhibitory dopaminergic pathway connecting the thalamus and the frontal lobe. Despite the
laughter, which has been incorrectly termed euphoria, many patients are depressed
and are embarrassed by the inability to control this often incongruous
response.
Depression
Depression,
or bipolar affective disorder, is clearly associated with multiple sclerosis
and may precede the onset of symptoms of multiple sclerosis in some cases.
Character or personality changes with impulsiveness or less inhibition in social
interactions may present problems or embarrassment to family members.
Sexual
Dysfunction
Sexual
dysfunction is not uncommon in both men and women with multiple sclerosis. Men
experience difficulty in achieving an erection because of diminished penile
sensation or difficulty maintaining an erection. Others report failure of
orgasm. Sexual dysfunction in women with multiple sclerosis includes lower limb
spasticity, lack of vaginal lubrication,
and diminished vaginal sensation, any of which can interfere with sexual
functioning.
Seizures
Epilepsy
occurs in 1 to 5 percent of patients with multiple sclerosis, a higher
frequency than in the normal population. Seizures are associated with lesions
in the cortical or subcortical area and the
onset is usually associated with the presence of new lesions in the cortical
gray matter or in subcortical regions.
When seizures are associated with clinical relapse, the seizures rarely recur.
Seizures not related to clinical relapse tend to recur occasionally but control
is usually straightforward. Patients with multiple sclerosis, seizures, and
progressive cognitive decline have a poor prognosis and are susceptible to
status epilepticus.
Tonic Spasms
Tonic spasms
are paroxysmal, unilateral stereotypical spasms of short duration precipitated
by movement or hyperventilation, lasting
30 to 90 s and involving part or the whole of one side of the body. The attacks
may be heralded by brief clonic movements.
During an attack, the affected limb or limbs are usually extended, but the
hands, fingers, feet, and toes may be drawn into a pseudodystonic posture.
There may be a spread to the face on the same side with head turning. Speech
may be affected by the distortion of the face. The patient is fully alert and
usually experiences minimal pain or discomfort. The affected limbs may have a
slight degree of weakness after an attack. The condition is believed to be the
result of acute demyelination involving the corticospinal
tracts in the brainstem or spinal
cord.
Lhermitte's
Sign
Flexion of
the head may result in an electric-like shock passing down the spine and into
the limbs. This phenomenon, known as Lhermitte's sign, while not pathognomonic, is highly suggestive of
multiple sclerosis and may also precede the development of other symptoms by
months or years in some cases.
Spasticity
The majority
of patients with multiple sclerosis will show some evidence of spasticity, which may vary from a slight increase
in tone to severe spastic paraplegia in flexion with limbs held in a permanent
flexed posture at the knees and hips. In the mildest of cases, spasticity may present with no more than a
slight increase in tendon reflexes and extensor plantar responses. In the most
severe cases, spasticity may dominate the
clinical picture and be responsible for severe disability.
Psychiatric
Symptoms
Psychosis
can occur in both chronic, progressive and relapsing-remitting forms of
multiple sclerosis when it heralds increasing activity of the disease process.
Paranoia or hallucinations are unusual and are occasionally prominent symptoms
when there is extensive involvement of both frontal and temporal lobes.
Fatigue
The majority
of patients experience fatigue. The onset may be sudden and debilitating, with
inability to continue even the simplest of tasks. Fatigue tends to be provoked
by a high atmospheric temperature and many patients relate difficulties in
functioning in the summer. Some are extremely sensitive to heat and report
profound weakness after a hot bath or shower. A febrile illness has the same
effect, with the appearance of symptoms suggesting a relapse of the disease.
However, there is rapid return to the prefebrile state once the fever subsides.
Pain
Multiple
sclerosis is not a painless disease and pain is occasionally a prominent
feature. As many as 80 percent of patients experience painful muscle spasms,
intermittent or constant limb pain, or spinal pain. Primary pain is usually
dysesthetic, occurring most commonly in the lower limbs. However, truncal and upper limb dysesthesias can occur. The dysesthesias
may be augmented by tic-like pains, tonic seizures are occasionally
painful, and in some cases, Lhermitte's sign is experienced as pain rather than
paresthesias. Chronic pain can occur as a dysesthesia in the extremities, in
girdle-like fashion around the waist or abdomen, as low back pain or pain in
the shoulders due to disuse with capsular adhesions.
Trigeminal neuralgia or atypical facial
pain can occur at any stage of the disease. The occurrence of trigeminal neuralgia in a young person should
always arouse the suspicion of multiple sclerosis.
Debilitated
patients who use a wheelchair often develop joint pains from abnormal posture
or from propelling the wheelchair manually. Spasticity
and muscle cramps can cause severe pain.
Headache
Migraine
headaches are not unusual in multiple sclerosis. Retro-orbital pain presenting
as a dull ache, and increasing on eye movement, occurs in optic and retrobulbar neuritis.
Respiratory
Impairment
The
incidence of respiratory failure in multiple sclerosis is low and usually
occurs in the presence of extensive spinal cord or brainstem involvement. Clinical indications of impending
respiratory failure include orthopnea, paradoxical
movements of the chest wall and abdominal muscles during respiration, and use
of accessory muscles of respiration. Patients with suspected impairment of pulmonary
function should be monitored carefully with a number of pulmonary function
tests, including force vital capacity (FVC), maximal voluntary ventilation
(MVV), and maximal expiratory pressure (MEP), coupled with the index score for
pulmonary function, which provides a clinical tool for the rapid assessment of
significant respiratory dysfunction in multiple sclerosis.
Pregnancy
Multiple
sclerosis does not reduce fertility. An apparent reduction in fertility may be
secondary to physical disability and to counseling against pregnancy by
physicians. Other factors include a decision by women with multiple sclerosis
to forego marriage, to have fewer children, or to undergo sterilization.
Multiple sclerosis does not affect the course of pregnancy and there is no
difference in the duration of labor or frequency of difficult delivery,
premature labor or stillbirth.
Relapse
rates of multiple sclerosis during pregnancy are significantly reduced,
particularly in the third trimester, but there is a significant increase in
relapse rates during the first 3 months postpartum.
Consequently, these facts should be discussed with a woman seeking
advice prior to pregnancy and to her partner, explaining the present knowledge
concerning pregnancy and multiple sclerosis. It is important that the partner
realize that he will have to assume more responsibility for child care and
reduce the burden of responsibility on the mother.
There is an
apparent beneficial effect of pregnancy in autoimmune diseases such as
rheumatoid arthritis, suggesting that the beneficial effects of pregnancy
could be the result of immunomodulation or immunosuppression.
However, the explanation for the alteration in the immune system remains
illusive.
Diagnostic
Procedures
1. The diagnosis of multiple sclerosis is established
by careful interpretation of clinical signs and symptoms. These indicate
multiple levels of CNS involvement. The diagnosis may be strengthened by
interpretation of other findings discussed below, but diagnosis remains a matter
of clinical judgment.
2. All patients should be fully investigated
for the presence of infection. This includes aerobic and anaerobic blood
cultures, urinalysis, culture and sensitivity,
and chest x-ray to rule out pneumonia. Many patients have infections that are
latent or occult. This dramatically affects response to treatment unless infection
is eradicated. Patients with decubitus ulcers,
which are chronic and deep, should receive bone scans to rule out the presence
of osteomyelitis.
3. Magnetic resonance imaging (MRI). An MRI
scan of the brain is abnormal in 95 percent of definite cases of multiple
sclerosis, but abnormal MRI findings alone are not sufficient to confirm a diagnosis
of multiple sclerosis without compatible clinical abnormalities.60
MRI scans are abnormal in only 70 percent of patients with probable multiple
sclerosis and 30 to 50 percent of patients with possible multiple sclerosis,
and some patients with multiple sclerosis may have normal MRI findings.
When
patients have a diagnosis of probable multiple sclerosis, a positive MRI scan
will raise the category to definite in about 50 percent of cases. The results
of positive MRI scanning in those categorized as possibly having multiple
sclerosis are less impressive, with only 5 percent changing category from possible
to definite. Nevertheless, progression to definite multiple sclerosis is more
likely in those with disseminated MRI lesions at presentation and less likely
in those without disseminated lesions. Other characteristic features are
immediate proximity to the ventricles, lesions greater than
Frequent
interval MRI scans have shown that newly encountered lesions found on
T2-weighted images have a transient enhancement following administration of
gadolinium. In about two-thirds of the cases, these enhancing lesions will
continue to show faded enhancement for 4 to 6 weeks with less than 2 percent
showing enhancement beyond 3 months. Enhanced lesions tend to appear
in clusters over time and lesions seen in T2-weighted MRI scans can regress in
size but are unlikely to disappear. Serial studies have shown that the MRI
attack rate greatly exceeds the clinical attack rate. There is a much lower
rate of new lesions defined by MRI in primary progressive multiple sclerosis
than in secondary progressive multiple sclerosis and in the
relapsing-remitting form of the disease.
Serial
studies have also shown a considerable amount of clinically silent disease
activity in relapsing-remitting and secondary progressive multiple sclerosis,
but there is a lack of correlation between MRI and clinical disability.
Nevertheless, MRI as a measure of multiple sclerosis activity is now widely
accepted as a surrogate marker of disease in treatment trials of evolving
therapies.
- To put veridical MS
we have to reveal in patient at least 2 focuses of lesion and 2
exacerbations, or 2 exacerbations of 1 clinical focus and 1 paraclinical
supposed focus.
- According to the
accepted criteria there should be at least 3 focuses in MRI (2 of them
should be located paraventricularly, 1 – subtentorialy (that means in
brain stem or cerebellum). The diameter of focuses should be at least

4. Examination of the cerebrospinal fluid (CSF). Acute exacerbations of multiple
sclerosis may be accompanied by a lymphocytic or
polymorphonuclear pleocytosis in the
CSF. This is short-lived and does not usually exceed 200 cells per cubic
millimeter. The CSF protein is elevated, particularly in early cases and
during acute exacerbations. The level rarely exceeds 100 mg/dL. Gamma globulin
elevation is seen in many cases and exceeds 13 percent of the total protein
content. About 70 percent of patients have evidence of abnormal intrathecal IgG synthesis, as demonstrated by
the IgG index.
IgG CSF/IgG
serum albumin CSF/albumin serum
An index
greater than 0.7 indicates synthesis of IgG within the CNS. The presence of IgG
oligoclonal bands is a more sensitive measure of local IgG production. However,
this finding is nonspecific
and oligoclonal bands in the CSF have been seen in patients suffering
from cerebral infarction, brain tumors, paraneoplastic
syndromes, diabetes mellitus, borreliosis, neurosyphilis, human immunodeficiency virus (HIV) infection,
various connective tissue diseases, and hypothyroidism.
Consequently, testing must include both serum and CSF. Detection in CSF
alone or primarily in the CSF is an indication of local IgG synthesis, which is
usually associated with multiple sclerosis. Oligoclonal bands (Table 7-2) are
detected in 90 percent of the patients with clinically definite multiple
sclerosis. This figure drops to 50 percent in optic neuritis and isolated brainstem and spinal cord disease. Elevation of
myelin basic protein is present in
approximately 80 percent of cases of acute multiple sclerosis or multiple
sclerosis in exacerbation. Antibodies to myelin,
myelin basic protein (MBP), myelin
oligodendrocyte glycoprotein (MOG), myelin-associated glycoprotein (MAG), and prote-olipid protein
(PLP) are present in cells in the CSF but are not specific for
multiple sclerosis.
5. Visual
evoked potentials are positive in about 80 percent of patients with multiple
sclerosis. Many of these patients have not had any visual symptoms. Similarly,
auditory evoked potentials are positive in about 70 percent of patients with
multiple sclerosis and a number of these cases do not show clinical signs of brainstem involvement. Somatosensory evoked potentials are positive in about 60 percent
of cases of multiple sclerosis. The presence of abnormal evoked potentials
provide additional objective evidence of a
heterogenous involvement of the CNS.
Table 7-2
Oligoclonal
Bands
Not specific
for multiple sclerosis. Can occur in any disease with demyelination. Multiple
sclerosis Neurosyphilis Viral
encephalitis Bacterial meningitis Cerebral lupus erythematosus Lyme disease Neurosarcoidosis
6. Neuropsychological
testing is useful, particularly in the early stages of apparent
intellectual and cognitive failure, when it is necessary to distinguish
between early dementia and depression.
7. Urological evaluation. Patients with established
symptoms of urgency and frequency of micturition sufficient to cause
inconvenience should have a limited urological evaluation with measurement of
postvoid residual urine and a cystometrogram.
8. Ophthalmology evaluation.
An opinion from an
ophthalmologist should be obtained in the event of visual problems. Baseline
evaluations of visual acuity, visual fields, color vision and the presence of
scotomata are determined accurately and can be used for a comparison should
visual deterioration continue.
Differential
Diagnosis
Multiple
sclerosis can mimic almost any chronic disease affecting the CNS. The
diagnosis is usually not difficult in well established cases, with evidence of
multiple areas of involvement in the CNS, but early cases often present a
problem in diagnosis. Various conditions (Table 7-3) can be confused with
multiple sclerosis.
1. The leukodystrophies.
The adult forms of metachromatic leukodystrophy, Fabry disease, X-linked adrenaleukodystrophy, globoid
leukodystrophy, and leukodystrophy with
diffuse Rosenthal fiber formation can
present with progressive deterioration and evidence of multiple areas of
involvement in the CNS. There is a peripheral neuropathy with slowing of
nerve conduction velocities
in metachromatic leukodystrophy, which is not present in multiple sclerosis.
The demonstration of metachromatic material,
low levels of arylsulfatase, and very long-chain fatty acids will establish the
diagnosis. The diagnosis of leukodystrophy with
diffuse Rosenthal fiber formation can be
made only by brain biopsy.
2. Spinocerebellar
degenerations. Autosomal dominant spinocerebellar degenerations, sporadic late-onset olivopontocerebellar atrophies (multisystem disease), and
Friedrich's ataxia are occasionally misdiagnosed as multiple sclerosis. Diagnostic
procedures in late-onset ataxias include
MRI scanning of the brain and spinal cord and nerve conduction studies.
Metabolic evaluation consists of peripheral blood smears for acanthocytosis;
determination of plasma amino acids,
vitamin E, lactate and pyruvate levels; lipid and lipoprotein determination;
and urinary organic acid to identify abetalipoproteinemia,
hypobetalipoproteinemia, vitamin E dysmetabolism, mitochondrial cytopathies, and organic acidemias.
Table 7-3
MRI-Detected
Abnormalities in White Matter Resembling Changes in Multiple Sclerosis
Acute
disseminated encephalomyelitis
Adult-onset leukodystrophies
Multisystem
disease
Spinocerebellar degeneration
Closed head
injury
HIV
encephalitis
HTLV-1
myelitis
Progressive multifocal leukoencephalopathy
Neurobrucellosis
Chronic granulomas
Beh§et
disease
Sjogren syndrome
Brain
tumors
Lymphoma
Cerebrovascular disease
Migraine
ischemia
Cerebral vasculitis
B12
deficiency
Moyamoya
Aging
Drug-induced
encephalopathy
Cerebral
lupus erythematosus
Neurosarcoidosis
Effects of
radiation therapy
3. Syphilis.
Both meningeal and vascular syphilis may mimic multiple
sclerosis. The diagnosis is established by abnormalities in the CSF and a positive
serologic test for syphilis.
4. Wilson disease (hepatolenticular degeneration) usually presents with symptoms and
signs of hepatic involvement and
neurological dysfunction.
However, a number of patients have minimal hepatic involvement with a broad
range of neurological signs, including dysarthria,
involuntary movements, and deteriorating coordination, followed by
progressive dementia and behavioral abnormalities. A misdiagnosis of multiple sclerosis, particularly in those with
marked cerebellar ataxia or psychiatric
disorder, is not uncommon. Diagnosis is established by a positive slit lamp
examination for Kayser-Fleischer rings, cataracts, and determination of serum,
copper and ceruloplasmin, and of 24-hour urinary excretion of copper.
5. The
antiphospholipid syndrome, which usually presents with deep venous
thrombosis or stroke, can mimic multiple sclerosis when repeated minor strokes
produce focal deficits and optic neuritis in a young adult.74
Moreover, some patients will show the presence of areas of increased signal
intensity in the periventricular area on
a T2-weighted MRI. The diagnosis of the antiphospholipid syndrome is
established by a positive anticardiolipin and lupus anticoagulant in the serum.
6. Lyme disease
is of major concern in the eastern United States and appears to be spreading
south and west. The disease is the cause of intermittent neurological
symptoms. In the mildest form, it causes
Bell's palsy, but
there is a
chronic encephalomyelitis with intermittent signs of CNS involvement.
There may be
a history of tick bite followed by a migratory rash and arthralgias. Lyme titers or Lyme
polymerase chain reaction in the blood or spinal fluid will be positive
in these patients.
Theme: Demyelination diseases of nervous system: multiply sclerosis (MS), acute
disseminated encephalomyelitis (ADEM), optic encephalomyelitis,
encephalomyelopolyradiculoneuritis, polyencephalomyelitis, disseminated myelitis. Acute myelitis.
Multiple
sclerosis is a demyelinating disease of the central nervous system (CNS) caused
by an autoimmune reaction that is the result of a complex interaction of
genetic and environmental factors.
Epidemiology
Multiple
sclerosis is more common in women, with a female:male ratio of 2.1:1. The
disease is rare in children and has a peak incidence at 30 to 33 years of age,
with a decline in the late forties. The upper limit of age at onset has usually
been accepted as 59 years, but late onset of multiple sclerosis after the age
of 60 years is reported. However, these cases probably represent late
occurrence of overt symptoms in individuals who have had the disease in a subclinical or unrecognized fashion for many
years.
The natural
history of multiple sclerosis has been studied extensively and it is clearly a
disease of temperate zones, with an increase in the prevalence gradient south
to north in the northern hemisphere, and north to south in the southern
hemisphere. Three zones of high, medium, and low prevalence rates can be recognized.
High-frequency prevalence rates of more than 30 per 100,000 population occur in
areas lying between latitudes 45° and 65° north or south. This includes
northern Europe, southern
These figures
cannot be attributed to climate alone, however, because there are
well-documented ethnic differences. Multiple sclerosis predominantly affects
people of northern European extraction. In the
Such
findings suggest a genetic factor in multiple sclerosis, and family studies
have demonstrated that the risk of multiple sclerosis is increased for relatives
of patients with the disease. Studies in
At the
present time, evidence suggests that multiple sclerosis is influenced by
several genes, the major histocompatibility (MHC)
complex class 2, HLA-DRII allele having
the strongest association with the disease in northern European populations.
However, the association of different class 1 and class 2 alleles has been reported in other populations.
Consequently, the current impression is that multiple sclerosis is probably polygenic, the result of complex genetic
factors involving the interaction of genes and coding within and outside of the
MHC complex. However, epidemiologic studies
in
At the
present time, it is considered probable that both genetic and environmental
factors are involved in multiple sclerosis, with infection as the major
environmental agent, in that both viral and bacterial infections can initiate
or precipitate attacks of multiple sclerosis. Evidence for a direct involvement
of a viral agent such as human T-cell lymphotropic virus (HTLV)-l, herpes
simplex virus (HSV)-l, HSV-6, scrapie,
parainfluenza virus 1, measles virus, coronavirus, simian virus,
chimpanzee cytomegalovirus, and LM7 retrovirus in multiple sclerosis is less
compelling. The role of environmental factors other than infection has been
studied, including the relationship of trauma and multiple sclerosis,
indicating that patients are at no greater risk to experience an exacerbation
of multiple sclerosis after trauma than at other times, nor is it likely that
trauma is ever a causal factor in initiating the disease process. The role of
emotional stress in multiple sclerosis is more controversial because stress is
difficult to define and quantitate.
Etiology,
Pathology, and Pathogenesis
The etiology
is unknown. The pathological changes in multiple sclerosis show variation,
depending on the age of focal demyelination (the plaque). In the acute state,
there is active demyelination with accumulation of sudanophilic myelin breakdown products. The area is edematous and there is marked perivascular cuffing around veins and venules by lymphocytes and macrophages. Plaques are frequently located in
the periventricular distribution,
particularly in the cerebral hemispheres, but plaques can occur at any site in
the white matter and often penetrate into the gray matter of the cortex and
deeper gray matter structures in the cerebrum and cerebellum. Because multiple
sclerosis is a disease of the CNS, plaque formation is not uncommon in the brainstem, cerebellum, spinal cord, and the
optic nerves, which are structurally part of the CNS, in which the oligodendrocytes are anti-genically similar to
the oligodendrocytes in the spinal cord.
All multiple
sclerosis lesions show a variable degree of axonal
loss, ranging from 10 to 20 percent in milder forms of the disease, to
80 percent in severe, acute multiple sclerosis.
Epidemiologic studies support the concept
that multiple sclerosis results from an aberrant immune reactivity occurring in
a genetically susceptible host who has acquired a specific, or one or more
nonspecific, neurotropic infections at a
critical age. Genetic susceptibility is thought to be associated with genes
within or close to the HLA-DR DQ subregion, located in the short arm of
chromosome 6. This primary infection results in a self-sustaining,
organ-specific autoimmune disorder that remains latent until activated by a subsequent
infection years after the primary event. An alternative explanation suggests
that the mechanism is one of persistent systemic viral infection, which
contributes to the changes in the CNS periodically, or to a persistent CNS
viral infection that is targeted by T cells unpredictably, resulting in an
inflammatory response producing myelin damage
(bystander response). The common factor in any one of these theoretical
situations is the activation of an autoimmune event within the CNS, directed
against myelin antigen-specific T cells,
but no specific antigen has as yet been identified. Antibodies to myelin basic protein appear to be the most
frequent finding in multiple sclerosis, but reactivity to other myelin antigens is a possibility. Candidates
include proteolipid protein, the most abundant myelin
protein in humans, myelin oligodendrocyte
glycoprotein, myelin-associated protein, minor myelin proteins, or heat shock proteins.
The
autoimmune reaction probably begins with a systemic infection that is associated
with liberation of γ-interferon, resulting in activation of CD4 T lymphocytes.
The lymphocytes attach to adhesion molecules on the surface endothelium of postcapillary
venules, roll along the surface of the endothelium,
producing endothelial cell
activation, followed by passage of the CD4 T lymphocytes into the CNS—in
effect, disrupting the blood-brain barrier. Once within the CNS, the
T-lymphocyte receptors respond to antigen presented by MHC class 2 molecules on
macrophages and astrocytes, but oligodendrocytes are
usually preserved at this stage. The antigen T-cell receptor interaction is
followed by stimulation of helper T cells, T-cell proliferation, and B-cell and
macrophage activation, with release of cytokines such as •γ-interferon, tissue
necrosis factor, interleukin-12, and proteases. The cytokines induce a local inflammatory reaction with further
disruption of the blood-brain barrier, followed by a major influx of CD4
lymphocytes and monocytes into the
lesion. Myelin damage results
from the combined effect of cytotoxic cytokines,
particularly tissue necrosis factor and cytotoxic
cells.
Oligodendrocytes appear to
survive and proliferate in the presence of acute demyelination and interact
with hypertrophied astrocytes, an
association that may represent a short-term protective mechanism.
Consequently, active demyelination and remyelination can occur in acute
lesions. Oligodendrocyte depletion in
chronic multiple sclerosis lesions may be a slow, insidious process, spanning a
protracted period during which there is gradual cell dropout.
Classification of Multiple
Sclerosis
Although
multiple sclerosis can affect any site in the CNS, it is possible to recognize
eight types of the disease (Table 7-1):
1. Relapsing-remitting multiple sclerosis.
This is the classical form of multiple sclerosis that often begins in the late
teens or twenties with a severe attack followed by complete or incomplete
recovery. Approximately 70 percent of patients with multiple sclerosis
experience a relapsing-remitting course initially.38 Further
attacks occur at unpredictable intervals, each followed by increasing
disability. The relapsing-remitting pattern tends to change into the secondary
progressive form of the disease in the late thirties.
2. Primary progressive multiple sclerosis. The
disease runs a steady deteriorating course that may be interrupted by periods
of quiescence without improvement. The rate of progression is variable; at its
most severe, this form of multiple sclerosis can terminate in death within a
few years. In contrast, the more chronic form of progressive multiple sclerosis
is similar to the benign form of the disease.
Table 7-1
Eight Types
of Multiple Sclerosis
1. Relapsing-remitting
2. Primary progressive
3. Secondary progressive
4. Relapsing progressive
5. Benign
6. Spinal form
7. Neuromyelitis
optica (Devic disease)
8. Marburg variant
3. Secondary progressive multiple sclerosis.
The relapsing-remitting form of the disease frequently develops into secondary
progressive multiple sclerosis after a variable period of time but usually in
the late thirties.
4. Relapsing progressive multiple sclerosis.
Occasional cases are encountered where patients with a progressive form of
multiple sclerosis have superimposed relapses with no significant recovery.
5. Benign multiple sclerosis. About 20 percent
of cases have the benign form of multiple sclerosis. This may be defined as
multiple sclerosis in which the patient is able to function at the level of
full employment or provide care of home and family independently 10 years after
the appearance of the first symptoms. It is extremely unlikely that these patients
will ever be incapacitated by the disease and they should continue to live a
full life span with only occasional minor symptoms.
The
existence of a benign form of multiple sclerosis increases the importance of
recording the date of the first symptoms in patients who appear to have few
residual abnormal signs several years after the onset of the disease. These patients
may be informed that they have a benign form of multiple sclerosis 10 years
following their first recorded symptom and that the benign course will continue
in the years ahead.
6. Spinal form of multiple sclerosis. This form of multiple sclerosis presents with
symptoms and signs of predominantly spinal cord involvement from the beginning
and maintains this pattern. There may be a clear-cut pattern of relapse and
remission initially, followed by the secondary progressive form of the disease
after several years, or the presentation may be one of steady deterioration
from the onset.
7. Neuromyelitis optica (Devic syndrome). Most cases of
this syndrome are believed to be examples of multiple sclerosis presenting
with acute transverse myelitis followed by optic neuritis. Many patients
follow a relapsing-remitting course indistinguishable from multiple sclerosis.
8. Marburg variant. This rare and malignant
form of multiple sclerosis is associated with a fulminating course of
progressive impairment of consciousness, severe visual loss, dysarthria, dysphagia, respiratory
insufficiency, and rapid deterioration. It is indistinguishable from acute
disseminated encephalomyelitis. The
Marburg variant may result from the autoimmune process of multiple sclerosis
occurring in an individual
with developmentally immature myelin
basic protein.
Clinical
Features
The
diagnosis of multiple sclerosis is based on the clinical demonstration of
multiple levels of involvement of the CNS. Symptoms may be grouped under
several headings.
Sensory
Symptoms
Sensory
symptoms are the most common symptoms experienced by patients with multiple
sclerosis. These symptoms are often forgotten or ignored by both patient and
physician. Even prolonged sensory symptoms fail to evoke concern, in contrast
to the almost immediate response that occurs with weakness or paralysis.
Consequently, many patients date the onset of multiple sclerosis from the first
appearance of weakness, visual loss, or other symptoms of dramatic onset rather
than forgotten or poorly recorded sensory symptoms.
Sensory
symptoms include impairment of sensation (hypesthesia), tingling (paresthesias), and uncomfortable sensations (dysesthesias) often referred to as
"burning," which may be present for days, weeks, or months without
objective abnormalities. All patients with suspected multiple sclerosis should
be carefully questioned about the occurrence of previous sensory symptoms.
Motor
Symptoms
Paralysis or
paresis of upper or lower limbs is the most common presenting symptom in
patients with multiple sclerosis. Paraparesis is
a common early complaint when the patient gives a history of increasing
weakness and stiffness of the lower extremities, associated with progressive
impairment of gait. Examination shows signs of upper motor neuron involvement
with spasticity, increased tendon
reflexes, and extensor plantar responses. These findings may be quite subtle in
the early states of the disease.
Visual Symptoms
Optic
neuritis presents with sudden visual loss and pain on eye movement and a
unilateral headache. The condition may be followed by a rapid progression to
total loss of vision in the affected eye (Fig. 7-1) When vision is preserved,
there is monocular blurring; a central, paracentral,
or centrocecal scotoma; and
impairment of color vision. In optic neuritis, there may be edema of the optic
discs, but the appearance can be normal in retrobulbar
neuritis, when the inflammatory response is localized and located
proximally in the optic nerve. Temporal pallor is a later development because
of demyelination of the maculopapular bundle. However, subclinical involvement of the optic nerve can occur and may be
present without symptoms, when the patient first presents with signs of
multiple sclerosis or may be identified when the patient presents with the
first symptoms of optic neuritis, indicating prior subclinical involvement of the optic nerve.
Recovery
from optic neuritis is unpredictable. Many patients experience no further
problems for several years and then develop symptoms of brain or spinal cord
involvement, indicating multiple sclerosis. However, not all optic neuritis is
multiple sclerosis, and only 50 percent of adolescents and young adults who
present with the sudden onset of optic neuritis subsequently develop
multiple sclerosis.
Those with a
large time interval between the optic neuritis and the development of
additional symptoms have a better prognosis. The clinician should always
inquire about the possibility of preceding symptoms in any patient who presents
with optic neuritis because the presence of symptoms some years before the
more dramatic visual symptoms might indicate a more benign prognosis.
Diplopia is indicative of third or
sixth nerve involvement in the brainstem. The
fourth nerve is rarely involved in isolation. Intemuclear ophthalmoplegia is pathognomonic of multiple sclerosis and rarely has another
etiology. Unilateral or bilateral Marcus Gunn pupil is often present in optic
or retrobulbar neuritis.
Bladder
Involvement
The early
symptoms of bladder dysfunction consist of occasional urgency of micturition
followed by mild nocturia occurring once
a night. The events gradually increase in number, disturbing the sleep of the
patient and the bed partner. There is a concomitant increase in urinary
frequency and urgency during the day, ultimately resulting in incontinence.
Impaired bladder control is usually the result of spinal cord involvement in
multiple sclerosis and decreasing bladder control usually parallels increasing paraparesis. However, this is not an inevitable
relationship and some patients retain adequate bladder function even when paraplegic.
The
anatomical center for bladder control lies in the tegmentum of the pons. The
center is under the influence of a higher level of control located in the
medial aspect of the frontal lobes. Thus, the frontal lobes can signal the pontine bladder control center to inhibit
bladder function or to initiate bladder emptying at will or when convenient.
The pontine center then inhibits or
permits bladder contraction through connections that traverse the spinal cord
and exit through the parasympathetic outflow
in the S2-S4 sacral nerves supplying the bladder.
A number of
abnormal responses are associated with interruption of bulbar or spinal cord connections in multiple sclerosis.
1. Detrusor hyperreflexia.
The interruption of the afferent impulses from the detrusor muscle of the bladder to the pontine micturition center by spinal cord disease results in an
uninhibited reflex at the sacral cord level. The detrusor muscle is no longer inhibited as bladder volume increases
and detrusor contraction is initiated in
response to smaller volumes of urine, resulting in increasing frequency.
2. Detrusor sphincter dyssynergia. The normal
pattern of voiding is disturbed. The normal relaxation of the external
sphincter is impaired and detrusor contraction
is poorly coordinated and accompanied by contraction rather than relaxation of
the external sphincter. The result is poor flow of urine and incomplete
emptying of the bladder.
3. Detrusor hypocontractility is a failure to
empty the bladder secondary to insufficient detrusor
pressure or a fading contraction on voiding.
Cerebellar Symptoms
Tremor, dysarthria, truncal ataxia, and limb ataxia are frequent symptoms of multiple
sclerosis. Occasionally, cerebellar dysfunction
is the dominant feature in multiple sclerosis, when a patient presents with
adequate vision and muscle strength shows serious disability from the cumulative
effects of the several forms of cerebellar
ataxia.
Brainstem Symptoms
Many
patients with multiple sclerosis develop signs of brainstem involvement. Involvement of the oculomotor and sixth cranial
nerves as they traverse the substance of the brainstem
results in diplopia. Intemuclear ophthalmoplegia due to involvement of the
interaxial fibers connecting the third, fourth, and sixth nerve nuclei is not
uncommon. Sensory loss over the face indicates involvement of the afferent
fibers entering the pons from the trigeminal nucleus. Facial weakness may be due
to involvement of the seventh nerve in the pons.
Episodic dysarthria and dysphagia indicate involvement of the vagus
nerve in the medulla, and dysarthria may
be due to involvement of the vagus nerve, the glossopharyngeal
nerve, and the hypoglossal nerve
as they course through the medulla. Involvement of the corticospinal tracts in the brainstem
can produce a progressive spastic quadriparesis; involvement of the cerebellar connections results in limb and truncal ataxia.
Spinal Cord
Symptoms
Most
patients with established multiple sclerosis have signs of spinal cord
involvement. These signs include some degree of spastic paraparesis with increased tone in both lower limbs, bilateral
ankle clonus, increased tendon reflexes, and bilateral extensor plantar
responses. It is not unusual to see a progression of paraparesis with increasing disability. This does not necessarily
indicate progression of the disease but may be due to progressive gliosis of plaques in the spinal cord. This
scarring produces increasing traction on and destruction of axons descending from higher centers in the CNS
and results in increasing spasticity and paraparesis.
Abnormal
Bowel Function
Constipation
may be a major problem in advanced multiple sclerosis. Bowel incontinence can
be a devastating experience to a patient with multiple sclerosis, particularly
if the loss of control occurs in a social situation or in a crowded area such
as a shopping center. Many patients react to the incident with reluctance to
leave home and are extremely apprehensive if they do so.
Memory
Deficits and Dementia
Impaired cognitive
processing is not unusual in multiple sclerosis when patients often display a
verbal working memory deficit owing to a central processing problem. This has a
significant impact on reading or other tasks that require the maintenance of
verbal information over a short period of time. Dementia occurs in approximately
50 percent of cases, with less cognitive impairment in patients with
relapsing-remitting disease than in those with the progressive form. The
disease can, however, remain predominantly spinobulbar in form, with little
involvement of the white matter in the cerebral hemispheres and preservation of
intellect. Patients with demyelination in the periventricular
white matter of the brain often show an explosive emotional response
with inappropriate laughter or occasional crying during conversation. This
condition results from the interruption of an inhibitory dopaminergic pathway connecting the thalamus and the frontal lobe. Despite the
laughter, which has been incorrectly termed euphoria, many patients are depressed
and are embarrassed by the inability to control this often incongruous
response.
Depression
Depression,
or bipolar affective disorder, is clearly associated with multiple sclerosis
and may precede the onset of symptoms of multiple sclerosis in some cases.
Character or personality changes with impulsiveness or less inhibition in social
interactions may present problems or embarrassment to family members.
Sexual
Dysfunction
Sexual
dysfunction is not uncommon in both men and women with multiple sclerosis. Men
experience difficulty in achieving an erection because of diminished penile
sensation or difficulty maintaining an erection. Others report failure of
orgasm. Sexual dysfunction in women with multiple sclerosis includes lower limb
spasticity, lack of vaginal lubrication,
and diminished vaginal sensation, any of which can interfere with sexual
functioning.
Seizures
Epilepsy
occurs in 1 to 5 percent of patients with multiple sclerosis, a higher
frequency than in the normal population. Seizures are associated with lesions
in the cortical or subcortical area and the
onset is usually associated with the presence of new lesions in the cortical
gray matter or in subcortical regions.
When seizures are associated with clinical relapse, the seizures rarely recur.
Seizures not related to clinical relapse tend to recur occasionally but control
is usually straightforward. Patients with multiple sclerosis, seizures, and
progressive cognitive decline have a poor prognosis and are susceptible to
status epilepticus.
Tonic Spasms
Tonic spasms
are paroxysmal, unilateral stereotypical spasms of short duration precipitated
by movement or hyperventilation, lasting
30 to 90 s and involving part or the whole of one side of the body. The attacks
may be heralded by brief clonic movements.
During an attack, the affected limb or limbs are usually extended, but the
hands, fingers, feet, and toes may be drawn into a pseudodystonic posture.
There may be a spread to the face on the same side with head turning. Speech
may be affected by the distortion of the face. The patient is fully alert and
usually experiences minimal pain or discomfort. The affected limbs may have a
slight degree of weakness after an attack. The condition is believed to be the
result of acute demyelination involving the corticospinal
tracts in the brainstem or spinal
cord.
Lhermitte's
Sign
Flexion of
the head may result in an electric-like shock passing down the spine and into
the limbs. This phenomenon, known as Lhermitte's sign, while not pathognomonic, is highly suggestive of
multiple sclerosis and may also precede the development of other symptoms by
months or years in some cases.
Spasticity
The majority
of patients with multiple sclerosis will show some evidence of spasticity, which may vary from a slight increase
in tone to severe spastic paraplegia in flexion with limbs held in a permanent
flexed posture at the knees and hips. In the mildest of cases, spasticity may present with no more than a
slight increase in tendon reflexes and extensor plantar responses. In the most
severe cases, spasticity may dominate the
clinical picture and be responsible for severe disability.
Psychiatric
Symptoms
Psychosis
can occur in both chronic, progressive and relapsing-remitting forms of
multiple sclerosis when it heralds increasing activity of the disease process.
Paranoia or hallucinations are unusual and are occasionally prominent symptoms
when there is extensive involvement of both frontal and temporal lobes.
Fatigue
The majority
of patients experience fatigue. The onset may be sudden and debilitating, with
inability to continue even the simplest of tasks. Fatigue tends to be provoked
by a high atmospheric temperature and many patients relate difficulties in
functioning in the summer. Some are extremely sensitive to heat and report
profound weakness after a hot bath or shower. A febrile illness has the same
effect, with the appearance of symptoms suggesting a relapse of the disease.
However, there is rapid return to the prefebrile state once the fever subsides.
Pain
Multiple
sclerosis is not a painless disease and pain is occasionally a prominent
feature. As many as 80 percent of patients experience painful muscle spasms,
intermittent or constant limb pain, or spinal pain. Primary pain is usually
dysesthetic, occurring most commonly in the lower limbs. However, truncal and upper limb dysesthesias can occur. The dysesthesias
may be augmented by tic-like pains, tonic seizures are occasionally
painful, and in some cases, Lhermitte's sign is experienced as pain rather than
paresthesias. Chronic pain can occur as a dysesthesia in the extremities, in
girdle-like fashion around the waist or abdomen, as low back pain or pain in
the shoulders due to disuse with capsular adhesions.
Trigeminal neuralgia or atypical facial
pain can occur at any stage of the disease. The occurrence of trigeminal neuralgia in a young person should
always arouse the suspicion of multiple sclerosis.
Debilitated
patients who use a wheelchair often develop joint pains from abnormal posture
or from propelling the wheelchair manually. Spasticity
and muscle cramps can cause severe pain.
Headache
Migraine
headaches are not unusual in multiple sclerosis. Retro-orbital pain presenting
as a dull ache, and increasing on eye movement, occurs in optic and retrobulbar neuritis.
Respiratory
Impairment
The
incidence of respiratory failure in multiple sclerosis is low and usually
occurs in the presence of extensive spinal cord or brainstem involvement. Clinical indications of impending
respiratory failure include orthopnea, paradoxical
movements of the chest wall and abdominal muscles during respiration, and use
of accessory muscles of respiration. Patients with suspected impairment of pulmonary
function should be monitored carefully with a number of pulmonary function
tests, including force vital capacity (FVC), maximal voluntary ventilation
(MVV), and maximal expiratory pressure (MEP), coupled with the index score for
pulmonary function, which provides a clinical tool for the rapid assessment of
significant respiratory dysfunction in multiple sclerosis.
Pregnancy
Multiple
sclerosis does not reduce fertility. An apparent reduction in fertility may be
secondary to physical disability and to counseling against pregnancy by
physicians. Other factors include a decision by women with multiple sclerosis
to forego marriage, to have fewer children, or to undergo sterilization.
Multiple sclerosis does not affect the course of pregnancy and there is no
difference in the duration of labor or frequency of difficult delivery,
premature labor or stillbirth.
Relapse
rates of multiple sclerosis during pregnancy are significantly reduced,
particularly in the third trimester, but there is a significant increase in
relapse rates during the first 3 months postpartum.
Consequently, these facts should be discussed with a woman seeking
advice prior to pregnancy and to her partner, explaining the present knowledge
concerning pregnancy and multiple sclerosis. It is important that the partner
realize that he will have to assume more responsibility for child care and
reduce the burden of responsibility on the mother.
There is an
apparent beneficial effect of pregnancy in autoimmune diseases such as
rheumatoid arthritis, suggesting that the beneficial effects of pregnancy
could be the result of immunomodulation or immunosuppression.
However, the explanation for the alteration in the immune system remains
illusive.
Diagnostic
Procedures
1. The diagnosis of multiple sclerosis is established
by careful interpretation of clinical signs and symptoms. These indicate
multiple levels of CNS involvement. The diagnosis may be strengthened by
interpretation of other findings discussed below, but diagnosis remains a matter
of clinical judgment.
2. All patients should be fully investigated
for the presence of infection. This includes aerobic and anaerobic blood
cultures, urinalysis, culture and sensitivity,
and chest x-ray to rule out pneumonia. Many patients have infections that are
latent or occult. This dramatically affects response to treatment unless infection
is eradicated. Patients with decubitus ulcers,
which are chronic and deep, should receive bone scans to rule out the presence
of osteomyelitis.
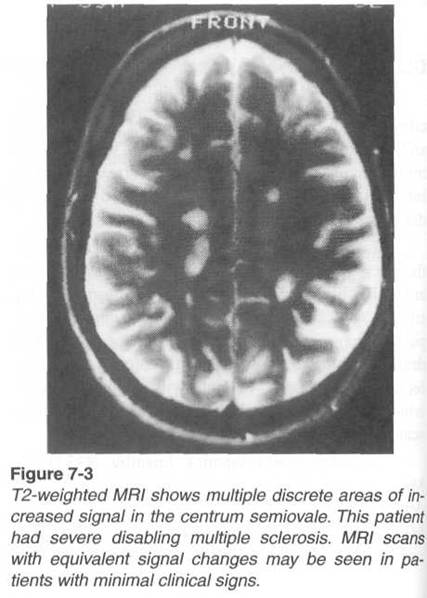
3. Magnetic resonance imaging (MRI). An MRI
scan of the brain is abnormal in 95 percent of definite cases of multiple
sclerosis, but abnormal MRI findings alone are not sufficient to confirm a diagnosis
of multiple sclerosis without compatible clinical abnormalities.60
MRI scans are abnormal in only 70 percent of patients with probable multiple
sclerosis and 30 to 50 percent of patients with possible multiple sclerosis,
and some patients with multiple sclerosis may have normal MRI findings.
When
patients have a diagnosis of probable multiple sclerosis, a positive MRI scan
will raise the category to definite in about 50 percent of cases. The results
of positive MRI scanning in those categorized as possibly having multiple
sclerosis are less impressive, with only 5 percent changing category from possible
to definite. Nevertheless, progression to definite multiple sclerosis is more
likely in those with disseminated MRI lesions at presentation and less likely
in those without disseminated lesions. Other characteristic features are
immediate proximity to the ventricles, lesions greater than
Frequent
interval MRI scans have shown that newly encountered lesions found on
T2-weighted images have a transient enhancement following administration of
gadolinium. In about two-thirds of the cases, these enhancing lesions will
continue to show faded enhancement for 4 to 6 weeks with less than 2 percent
showing enhancement beyond 3 months. Enhanced lesions tend to appear
in clusters over time and lesions seen in T2-weighted MRI scans can regress in
size but are unlikely to disappear. Serial studies have shown that the MRI
attack rate greatly exceeds the clinical attack rate. There is a much lower
rate of new lesions defined by MRI in primary progressive multiple sclerosis
than in secondary progressive multiple sclerosis and in the
relapsing-remitting form of the disease.
Serial
studies have also shown a considerable amount of clinically silent disease
activity in relapsing-remitting and secondary progressive multiple sclerosis,
but there is a lack of correlation between MRI and clinical disability.
Nevertheless, MRI as a measure of multiple sclerosis activity is now widely
accepted as a surrogate marker of disease in treatment trials of evolving
therapies.
- To put veridical MS
we have to reveal in patient at least 2 focuses of lesion and 2
exacerbations, or 2 exacerbations of 1 clinical focus and 1 paraclinical
supposed focus.
- According to the
accepted criteria there should be at least 3 focuses in MRI (2 of them
should be located paraventricularly, 1 – subtentorialy (that means in
brain stem or cerebellum). The diameter of focuses should be at least
4. Examination of the cerebrospinal fluid (CSF). Acute exacerbations of multiple
sclerosis may be accompanied by a lymphocytic or
polymorphonuclear pleocytosis in the
CSF. This is short-lived and does not usually exceed 200 cells per cubic
millimeter. The CSF protein is elevated, particularly in early cases and
during acute exacerbations. The level rarely exceeds 100 mg/dL. Gamma globulin
elevation is seen in many cases and exceeds 13 percent of the total protein
content. About 70 percent of patients have evidence of abnormal intrathecal IgG synthesis, as demonstrated by
the IgG index.
IgG CSF/IgG
serum albumin CSF/albumin serum
An index
greater than 0.7 indicates synthesis of IgG within the CNS. The presence of IgG
oligoclonal bands is a more sensitive measure of local IgG production. However,
this finding is nonspecific
and oligoclonal bands in the CSF have been seen in patients suffering
from cerebral infarction, brain tumors, paraneoplastic
syndromes, diabetes mellitus, borreliosis, neurosyphilis, human immunodeficiency virus (HIV) infection,
various connective tissue diseases, and hypothyroidism.
Consequently, testing must include both serum and CSF. Detection in CSF
alone or primarily in the CSF is an indication of local IgG synthesis, which is
usually associated with multiple sclerosis. Oligoclonal bands (Table 7-2) are
detected in 90 percent of the patients with clinically definite multiple
sclerosis. This figure drops to 50 percent in optic neuritis and isolated brainstem and spinal cord disease. Elevation of
myelin basic protein is present in
approximately 80 percent of cases of acute multiple sclerosis or multiple
sclerosis in exacerbation. Antibodies to myelin,
myelin basic protein (MBP), myelin
oligodendrocyte glycoprotein (MOG), myelin-associated glycoprotein (MAG), and prote-olipid protein
(PLP) are present in cells in the CSF but are not specific for
multiple sclerosis.
5. Visual
evoked potentials are positive in about 80 percent of patients with multiple
sclerosis. Many of these patients have not had any visual symptoms. Similarly,
auditory evoked potentials are positive in about 70 percent of patients with
multiple sclerosis and a number of these cases do not show clinical signs of brainstem involvement. Somatosensory evoked potentials are positive in about 60 percent
of cases of multiple sclerosis. The presence of abnormal evoked potentials
provide additional objective evidence of a
heterogenous involvement of the CNS.
Table 7-2
Oligoclonal
Bands
Not specific
for multiple sclerosis. Can occur in any disease with demyelination. Multiple
sclerosis Neurosyphilis Viral
encephalitis Bacterial meningitis Cerebral lupus erythematosus Lyme disease Neurosarcoidosis
6. Neuropsychological
testing is useful, particularly in the early stages of apparent
intellectual and cognitive failure, when it is necessary to distinguish
between early dementia and depression.
7. Urological evaluation. Patients with established
symptoms of urgency and frequency of micturition sufficient to cause
inconvenience should have a limited urological evaluation with measurement of
postvoid residual urine and a cystometrogram.
8. Ophthalmology evaluation.
An opinion from an
ophthalmologist should be obtained in the event of visual problems. Baseline
evaluations of visual acuity, visual fields, color vision and the presence of
scotomata are determined accurately and can be used for a comparison should
visual deterioration continue.
Differential
Diagnosis
Multiple
sclerosis can mimic almost any chronic disease affecting the CNS. The
diagnosis is usually not difficult in well established cases, with evidence of
multiple areas of involvement in the CNS, but early cases often present a
problem in diagnosis. Various conditions (Table 7-3) can be confused with
multiple sclerosis.
1. The leukodystrophies.
The adult forms of metachromatic leukodystrophy, Fabry disease, X-linked adrenaleukodystrophy, globoid
leukodystrophy, and leukodystrophy with
diffuse Rosenthal fiber formation can
present with progressive deterioration and evidence of multiple areas of
involvement in the CNS. There is a peripheral neuropathy with slowing of
nerve conduction velocities
in metachromatic leukodystrophy, which is not present in multiple sclerosis.
The demonstration of metachromatic material,
low levels of arylsulfatase, and very long-chain fatty acids will establish the
diagnosis. The diagnosis of leukodystrophy with
diffuse Rosenthal fiber formation can be
made only by brain biopsy.
2. Spinocerebellar
degenerations. Autosomal dominant spinocerebellar degenerations, sporadic late-onset olivopontocerebellar atrophies (multisystem disease), and
Friedrich's ataxia are occasionally misdiagnosed as multiple sclerosis. Diagnostic
procedures in late-onset ataxias include
MRI scanning of the brain and spinal cord and nerve conduction studies.
Metabolic evaluation consists of peripheral blood smears for acanthocytosis;
determination of plasma amino acids,
vitamin E, lactate and pyruvate levels; lipid and lipoprotein determination;
and urinary organic acid to identify abetalipoproteinemia,
hypobetalipoproteinemia, vitamin E dysmetabolism, mitochondrial cytopathies, and organic acidemias.
Table 7-3
MRI-Detected
Abnormalities in White Matter Resembling Changes in Multiple Sclerosis
Acute
disseminated encephalomyelitis
Adult-onset leukodystrophies
Multisystem
disease
Spinocerebellar degeneration
Closed head
injury
HIV
encephalitis
HTLV-1
myelitis
Progressive multifocal leukoencephalopathy
Neurobrucellosis
Chronic granulomas
Beh§et
disease
Sjogren syndrome
Brain
tumors
Lymphoma
Cerebrovascular disease
Migraine
ischemia
Cerebral vasculitis
B12
deficiency
Moyamoya
Aging
Drug-induced
encephalopathy
Cerebral
lupus erythematosus
Neurosarcoidosis
Effects of
radiation therapy
3. Syphilis.
Both meningeal and vascular syphilis may mimic multiple
sclerosis. The diagnosis is established by abnormalities in the CSF and a positive
serologic test for syphilis.
4. Wilson disease (hepatolenticular degeneration) usually presents with symptoms and
signs of hepatic involvement and
neurological dysfunction.
However, a number of patients have minimal hepatic involvement with a broad
range of neurological signs, including dysarthria,
involuntary movements, and deteriorating coordination, followed by
progressive dementia and behavioral abnormalities. A misdiagnosis of multiple sclerosis, particularly in those with
marked cerebellar ataxia or psychiatric
disorder, is not uncommon. Diagnosis is established by a positive slit lamp
examination for Kayser-Fleischer rings, cataracts, and determination of serum,
copper and ceruloplasmin, and of 24-hour urinary excretion of copper.
5. The
antiphospholipid syndrome, which usually presents with deep venous
thrombosis or stroke, can mimic multiple sclerosis when repeated minor strokes
produce focal deficits and optic neuritis in a young adult.74
Moreover, some patients will show the presence of areas of increased signal
intensity in the periventricular area on
a T2-weighted MRI. The diagnosis of the antiphospholipid syndrome is
established by a positive anticardiolipin and lupus anticoagulant in the serum.
6. Lyme disease
is of major concern in the eastern United States and appears to be spreading
south and west. The disease is the cause of intermittent neurological
symptoms. In the mildest form, it causes
Bell's palsy, but
there is a
chronic encephalomyelitis with intermittent signs of CNS involvement.
There may be
a history of tick bite followed by a migratory rash and arthralgias. Lyme titers or Lyme
polymerase chain reaction in the blood or spinal fluid will be positive
in these patients.
7. Behcet disease.
The presence of cranial neuropathies, cerebellar
ataxia, hemiparesis, quadri-paresis, pseudobulbar
palsy, and peripheral neuropathy in an individual with oral ulcers,
genital ulcers, and uveitis suggests Behcet disease. An MRI may reveal findings
resembling those seen in multiple sclerosis.
8. HTLV-1 infection. This retrovirus produces a myelopathy and spastic paraparesis
and is an occasional cause of more widespread white matter disease.
Under these circumstances, it may be difficult to distinguish HTLV-1 infection
from multiple sclerosis and appropriate antibody detection in both blood and
CSF is necessary.
9. Subacute combined
degeneration of the spinal cord may resemble multiple sclerosis, particularly
if there is an associated dementia and optic atrophy. Serum B12
levels are low in this condition. Although most cases of serum B12
deficiency are acquired, inherited defects have been described in infants and
children. Late-onset adult cases mimicking multiple sclerosis have occurred,
suggesting that patients diagnosed with multiple sclerosis should be screened
for B12 deficiency. Similarly, folate
dysme-tabolism in the rare hereditary adult presentation may resemble
multiple sclerosis.
10. Brain tumor. The presence of a fixed single
neurologic deficit in a young adult should always suggest the possibility of a
brain tumor rather than multiple sclerosis. The diagnosis is established by MRI
or computed tomography (CT) scanning.
11. The arteritides. Both polyarteritis nodosa and systemic lupus erythematosus can produce multiple lesions in
the CNS. However, other organs are often involved and there is evidence of
peripheral neuropathy with an elevated sedimentation rate, abnormal nerve
conduction velocities, and a positive nerve biopsy. Abnormal antibodies in
lupus erythematosus should reveal the
presence of this condition, but abnormal antibodies are not always present in
isolated cerebral lupus erythematosus. Similarly,
isolated cranial arteritis can mimic
multiple sclerosis and is often accompanied by a normal sedimentation rate and
a lack of any abnormal serum antibody levels. Diagnosis of this condition is established
by angiography, which reveals beading and
irregularity in the lumen of the intracranial arteries.
12. Transverse myelitis. Multiple sclerosis is
a relatively rare cause of transverse myelitis. Unless there is definite
evidence of multiple areas of involvement of the CNS, other conditions causing
transverse myelitis should be considered.
13. Mitochondrial
disorders. The association of optic atrophy, ataxia, spasticity, and hyperreflexia, usually associated with
multiple sclerosis, can occur in Leber's optic
atrophy, now believed to be the result of mitochondrial
DNA mutations. An electrocardiogram and molecular diagnostic tests are
suggested in suspected cases of Leber disease.
Other mitochondrial metabolic disorders can mimic
multiple sclerosis, including MELAS syndrome (mitochondrial
encephalomyopathy, lactic acidosis, and
stroke-like episodes), chronic milder forms of the MEERF syndrome (myoclonus epilepsy and ragged red fibers), and
the adult form of Leigh syndrome.
14. Sjogren syndrome.
There is a marked similarity in the clinical presentation of multiple sclerosis
and Sjogren syndrome with CNS
involvement. Both conditions may have optic neuritis, spinal cord involvement, psychiatric manifestations, abnormal evoked potentials, similar CSF
profiles, and indistinguishable abnormalities on MRI and CT scans. Features
that may distinguish Sjogren syndrome
include the sicca complex, xerophthalmia, xerostomia, or recurrent
salivary gland enlargement, peripheral neuropathy, vasculitis in skin or muscle, elevated sedimentation rate,
abnormal antinuclear antibody, positive rheumatoid factor,
anti-RO(SSA) or anti-LA(SSB) antibodies
and decreased complement.
15. Hereditary spastic paraparesis with progressive lower limb weakness and spasticity, hyperreflexia, and urinary
incontinence can mimic multiple sclerosis, particularly when there is an
associated optic atrophy. A positive family history suggesting an autosomal dominant trait and the slow
progression of the disease should lead to the exclusion of multiple sclerosis.
Treatment
1. Infection. Because there is strong evidence
that the first attack or exacerbation of multiple sclerosis may be caused by
viral or bacterial infection, all infections should be treated promptly.
2. Bed rest. Patients with acute multiple sclerosis
or an acute exacerbation of multiple sclerosis benefit from complete bed rest.
Patients who are removed from the necessity of self-care and added worries of
the home environment improve with rest. However, the period of rest should not
be protracted and no more than a few days in the great majority of patients.
Once the patient shows improvement, the institution of a graded program of
physical therapy becomes paramount.
3. Corticosteroids. Evidence suggests
that high doses of intravenous corticosteroids
(glucocorticoids) may arrest the progress of multiple sclerosis.
Corticosteroids have several
beneficial effects, including inhibition of secretion by antigen-presenting
cells and T cells of the cytokines, tumor
necrosis factor a, and interleukin-6. An additional effect may be inhibition
of secretion of 7-interferon and inter-leukin-12 by T cells.
About 85
percent of patients with relapsing-remitting multiple sclerosis shows objective
signs of neurological improvement during treatment with intravenous corticosteroids. Fifty percent of patients with
progressive multiple sclerosis are improved by intravenous corticosteroids, although for many, the
response is limited to a reduction in spasticity.
This figure may improve if attention is paid to prevention of infection
in the posttreatment period.
The
short-term use of intravenous corticosteroids is
attended by few side effects. Some patients experience insomnia during
treatment, a few show signs of euphoria; gastric upset with epigastric pain
responds to ranitidine 150 mg ql2h.
Hypomania or depression are unusual events. The daily dose and duration of
therapy have not been determined, but an intravenous dose of 1000 mg
methylprednisolone over 3 h daily for 7 days, followed by alternate-day oral
methylprednisolone, beginning with 96 mg (16 mg tablets X 6) at breakfast, and
reducing by 8 mg q.o.d., will allow the hypothalamic-pituitary-adrenal axis to
recover by the end of therapy. In the majority, this regimen will result in at
least 6 months' remission and several months or even years in many cases. The
treatment can be repeated if relapse occurs.
As indicated
under Diagnostic Procedures, all patients will have received full investigation
for infection, including blood cultures, urinalysis,
culture and sensitivity, and chest x-ray to rule out pneumonia before
receiving corticosteroid therapy.
Infections should be treated before and during treatment with corticosteroids. One of the major causes of
failure or poor response to corticosteroids is
the presence of a concomitant, untreated infection.
4. Immunosuppressive therapy. Long-term treatment
with immunosuppressants may reduce the
frequency of relapses in patients with multiple sclerosis. Azathioprine is
probably the safest drug in this category and has reduced relapse to 70 percent
in 3 years, compared to 80 percent in the placebo group.
Azathioprine
has few adverse effects and has not been shown to carry an increased risk of
inducing neoplasia, unlike the more
powerful immunosuppressive therapies
used to prevent transplant rejection.
Methotrexate, a drug used widely in the
treatment of chronic autoimmune diseases such as rheumatoid arthritis and
psoriasis, will reduce progression of disability in chronic progressive
multiple sclerosis. Low-dose therapy using 7.5 mg per week orally is effective,
and adverse effects are few, but regular monitoring with complete blood counts
and liver function tests is advised. Currently, low-dose oral methotrexate appears to be the best therapy for
slowing deterioration in chronic progressive multiple sclerosis.
Cladribine,
a specific antilymphocytic agent that is incorporated into DNA and induces
lymphocyte apoptosis, is reported to produce improvement in patients with
chronic progressive multiple sclerosis. The drug, which is
administered intravenously, has been reported to induce lymphopenia and severe but reversible aplastic anemia; the safety of cladribine has yet to be determined.
Results of
studies of cyclosporine in multiple sclerosis have been equivocal. The drug may
be beneficial in patients with frequently recurring or nearly continuous
disease activity but unacceptable toxicity and
marginal benefit have limited the use of cyclosporine in multiple sclerosis.
5. Total lymphoid
irradiation, originally introduced for the treatment of Hodgkin
disease, has theoretical benefits in multiple sclerosis, in that treatment
produces a significant reduction in CD4 and CD8 lymphocytes. Potential risks of
therapy, such as malignancy, have limited the use of this modality, and the
results of some studies have been equivocal.
6. Plasmapheresis has
marginal benefit in multiple sclerosis
and has not been accepted as an established therapy for this disease.
7. Interferon β.
Interferon β-lb (Betaseron) is
reportedly effective in reducing clinical attacks of multiple sclerosis by
approximately 30 percent over 24 months, when compared to placebo. Treatment
reduces frequency of major attacks by 50 percent and produces immediate and
significant reduction in contrast-enhanced MRI lesions and fewer new lesions
in patients receiving interferon B. The
dose is 8 million units subcutaneously every other day.
Interferon β-la (Avonex) is also an
available alternative therapeutic agent. It also lowers multiple sclerosis
attack frequency by 30 percent and decreases disease activity, measured by
gadolinium-enhanced MRI. The dose is 6 million units intramuscularly, once
weekly.
Adverse
effects of interferons include fever,
chills, headache, and myalgia. These "flu-like" symptoms begin 4 to
6 h after injection and last for a few hours. The response tends to resolve
after a few weeks of therapy but can persist for several months in a minority
of cases. Acetaminophen or ibuprofen, given 1 h before injection, reduces the
flu-like response. The dose of acetominophen or ibuprofen can be repeated,
should the flu-like symptoms still occur. The persistence of adverse effects
requires reduction of the dose by 50 percent, that is, 4 million units of
Betaseron and gradually increasing the dose to 8 million units over a period
of 4 weeks. Prednisone 20 mg given 2 h
before the injection of interferon 3 is also
effective in reducing adverse effects.
Other
adverse effects include redness at injection sites, which occurs in many
patients receiving Betaseron. The local reaction lasts for many weeks before
resolution. Necrosis at the injection site is rare, but persistence of painful
skin reactions requires the cessation of treatment.
Interferon β has been associated
with depression, which usually responds to a serotonin uptake inhibitor such
as fluoxetine. Inquiry should be made regarding
thoughts of suicide, which, if present, are an indication to stop treatment
with interferon p.
The
development of virus-neutralizing antibodies has been reported in 35 percent
of multiple sclerosis patients receiving interferon
β-lb. The appearance of antibodies has minimal effect on clinical
response and does not appear to be a reason for discontinuing therapy. The
action of interferon β in multiple
sclerosis appears to be multifactorial, including
sequestration of T lymphocytes into lymphoid tissue
and impaired migration of T lymphocytes through the blood-brain barrier by
inhibition of adhesion molecules on endothelial cells;
decreased release of cytokines, including
^-interferon, from T lymphocytes;
decreased tissue necrosis factor production by macrophages;
and paradoxically, increased inter-leukin-6 production.
8. Copolymer 1.
The development of copolymer 1 was based
on the premise that myelin basic protein
is encephalitogenic and can cause experimental allergic encephalomyelitis
(EAE) in animals. However, although some regions of the protein
are encephalitogenic, other regions will suppress the development of EAE. This
led to the synthesis and testing of several copolymers
of amino acids, based on the amino acid composition of myelin basic protein. One such copolymer, designated copolymer 1, suppressed EAE in guinea pigs and other animals. The
mechanism of suppression is not certain, but copolymer
1 seems to inhibit human T-cell lines specific for myelin basic protein. Consequently, the
application of copolymer 1 to multiple
sclerosis was a natural development
in therapy. A
double-blind placebo-controlled
trial of copolymer
9. Immunoglobulin
therapy. Treatment with intravenous immunoglobulin
has received some attention in recent years and there is some
indication that IVIG may be safe and effective in reducing the frequency of
exacerbations in relapsing-remitting multiple sclerosis.
10. Physical therapy. All patients with multiple
sclerosis should be evaluated by a physiatrist and should be placed in a graded
program of physical therapy. This program should be under constant review so
it can be modified, depending on the results of corticosteroid
and other therapies, as well as the benefit of the physical therapy
itself.
11. Spasticity.
Increased muscle tone, exaggerated tendon reflexes, clonus, and
spontaneous muscle spasms are often present in patients with advanced multiple
sclerosis. An acute increase in spasticity can
occur during an exacerbation of multiple sclerosis, or spasticity may present as an insidious deterioration over a period
of months, when the deterioration is often not apparent to patient or
therapist. Baclofen (Lioresal) is
effective in reducing spasticity and can
be given in doses up to 120 mg daily. The tendency is to underdose. The
medication should be given in an initial dose of 20 mg ql2h orally, with
gradual increments over several weeks to an effective level. High doses,
although reducing spasticity, may
increase weakness in some cases. Diazepam
(Valium) or clonazepam (Klonopin)
are potent spasmolytic agents. The tendency to drowsiness can be mitigated by
beginning with a low dose and only increasing when the patient is comfortable,
that is, not drowsy.
Dantrolene sodium (Dantrium) is effective
in reducing spasticity but has limited
applications because it almost invariably causes weakness. However, it can be
useful for treatment of spasticity in nonambulatory patients with severe prolonged
muscle contractions, who will not be adversely affected by the decrease in
voluntary muscle power associated with the use of this drug. Adverse effects
include damage to the liver, drowsiness, and light-headedness. An initial dose
of 25 mg/day may be increased by 25 mg increments every week, to a maximum
dose of 100 mg/day.
Tizanidine
is an effective antispastic agent with an antispasticity effect
comparable to baclofen. Tizanidine
dosage should be titrated beginning with 2 mg at night and gradually
increasing by 2 mg every 4 days in divided doses, until therapeutic goals have
been achieved without adverse effects. The larger dose should be given at
bedtime to minimize adverse effects. The average daily dose is 18 to 24 mg,
and the total daily dose should not exceed 36 mg. Adverse effects include dry
mouth, drowsiness, hypotension, light-headedness, abnormal liver function tests,
and the rare occurrence of hallucinations. These adverse effects tend to
decrease in intensity as the duration of therapy increases.
Sudden
muscle spasms (charley horses), whether nocturnal or diurnal, often respond to clonazepam, which is particularly useful for
nocturnal spasms, in that clonazepam not
only reduces spasm but also induces sleep without contributing to fatigue the
next day.
When
patients fail to respond to oral medication, intrathecal
baclofen delivered through a programmed pump placed in the abdominal
wall, with an intrathecal catheter in the
spinal canal, produces remarkable reduction of spasticity
and spasms in patients with severe spastic paraparesis. Potentially ambulatory patients have returned to
walking in some cases.
Botulinum toxin is effective in reducing
spasticity when injected into selected
muscles but has had limited application to date.
12. Visual difficulties. Patients should be encouraged
to report visual deterioration at the onset of the problem. Because optic neuritis can develop
rapidly, further deterioration should
be treated promptly, with
intravenous or oral corticosteroids in a
high dose. In many cases, this produces rapid improvement in symptoms or
stabilizes the condition, with subsequent slower but steady improvement in
visual acuity.
13. Weakness. It is very difficult to
strengthen a muscle weakened by
central denervation. The potassium channel blocking agents
4-aminopyridine and 3-4-diaminopyridine may improve action potential provocation
in demyelinated axons and improve
neurological function. Body cooling, using cooling vests or repeated cold
showers in the summer months, are effective in those who are heat sensitive.
14. Fatigue may strike without warning. Families
should be informed about the fatigue factor and the unpredictable development
of this symptom. This prevents resentment when the patient is suddenly unable
to attend a long-planned social function and when there is a need for extra
rest periods during the day. The intense fatigue associated with a febrile illness
will respond once the body core temperature returns to normal. Every patient
with recent onset of fatigue should be evaluated for depression, medication
effect, or intercurrent illness.
A number of
drugs may help eliminate fatigue, including amantadine
100 mg twice a day, pemoline (Cylert)
37.5 mg morning and noon, methylphenidate (Ritalin) 10
mg morning and
noon, fluoxetine (Prozac) 20 mg every morning, or selegiline 5 mg ql2h orally.
15. Pain is a common feature of multiple sclerosis.
The treatment consists of physical therapy, when appropriate, and medication.
Mild chronic pain may respond to acetaminophen or propoxyphene and acetaminophen
(Darvocet-N). Nonsteroidal
antiinflammatory drugs should be used with caution to avoid
gastric ulceration. Ibuprofen 600 mg,
given with meals, is an effective analgesic. Tramadol
(Ul-tram) 50 mg with misoprostol (Cytotec)
100 μg, to limit the risk of gastric ulceration,
will control moderately severe pain. Gabapentin beginning 300 mg 12h
and increasing, as tolerated, to as high as 2700 mg in divided doses, or
amitriptyline 10 mg q.h.s., increasing slowly by 10-mg increments to 80 to 100
mg daily, are both useful in pain control. Trigeminal
neuralgia usually responds to carbamazepine but the response is less
predictable in atypical facial pain. A painful Lhermitte's sign often shows
response to carbamazepine or clonazepam as
do tic-like extremity pains. If opioids are prescribed,
the medication should be
prescribed by one practitioner. The risk of addiction with oral opioids is low. but dependency can occur. Neurolytic nerve blocks are required occasionally,
including epidural blocks for chronic
sciatic pain.
16. Ankle edema. Swelling of the ankles in
patients with limited walking or in those who are nonambulatory and confined to a wheelchair is a gravity effect with
fluid accumulating in the dependent tissues of the feet and ankles.
Consequently, the use of diuretics is of little value. The patient should lie
supine for several hours a day, with the ankles elevated above the level of
the heart. This can be accomplished by having the patient lie supine with the
feet elevated on a firm cushion or over the armrest of a sofa, thus permitting
gravity-driven drainage over the lower limbs. The use of leotards or elastic
stockings may also be helpful.
17. Restricted mobility. Many patients with
limited mobility resist the use of a wheelchair and require a great deal of
persuasion to use an electric cart. The idea that this will lead to further
weakening is frequently expressed and is, of course, not true. The severe paraparetic with good upper limb function
should be encouraged to use an electric cart. The increased mobility and
broadening of the patient's social contacts is truly remarkable, once this is
accepted, and the electric cart is an essential therapeutic tool in many
cases.
18. Decubitus ulcers.
Skin care is one of the paramount needs in the wheelchair-bound paraplegic
patient or in those who are bedridden. With few exceptions, skin breakdown and
the development of decubitus ulcers is
the result of neglect by caregivers. Treatment
requires removal of pressure in the affected area and bacterial culture,
followed by the use of appropriate antibiotics when the ulcer is infected. When
the ulcer fails to heal or when the patient appears to be deteriorating, the
suspicion of an underlying osteomyelitis indicates the need for a radioactive
bone scan to confirm this diagnosis. A protracted course of intravenous
antibiotics is indicated in such cases. Deep (third degree) decubitus ulcers usually require debridement and plastic surgery with surgical
reconstruction.
19. Urinary tract infection. Cystitis is common
in female patients with multiple sclerosis and has an increased frequency in
male patients using self-catheterization. Although pyelonephritis is uncommon,
it can occur in severely debilitated patients and is a potent factor in chronic
illness, with anemia, weight loss, and fatigue. Urinary tract infections
require urinary culture and sensitivity testing, with the use of appropriate
antibiotic therapy. Attention to the symptoms of infection and immediate
treatment facilitates the use of a Foley catheter
or suprapubic catheter and increases the
safety of self-catheterization.
20. Management of bladder dysfunction.
A. Detrusor hyperreflexia. The early
symptoms of bladder dysfunction are usually caused by detrusor hyperreflexia, with urgency, frequency, and occasional nocturia. Most patients can be managed with an anticholinergic such as oxybutynin chloride 5
mg ql2h and increasing to 5 mg q8h if necessary. Tolterodine (Detrol) 2 mg ql2h
is equally effective and has fewer adverse effects. Imipramine PM 75 to 100 mg
q.h.s. is a useful alternative, particularly when nocturia is a problem. Pro-Banthine 15 mg with meals and at bedtime
is an effective alternative. Hyoscyamine time
release 0.375 mg at night is also effective in reducing nocturia.
B. Sphincter
detrusor dyssynergia with poor flow,
interrupted stream, and increased postvoid residual responds to prazosin 0.5 mg/day, increasing by 0.5-mg
increments to an effective dose, if there are no hypotensive effects. Doxazosin
mesylate tablets 1 mg q.h.s., increasing by increments to effect, are
also helpful.
C. Incomplete emptying with a post-micturition
residual volume greater than 150 mL
requires intermittent self-catheterization. This technique has revolutionized
the management of bladder dysfunction in multiple sclerosis but requires
instruction and is a clean rather than sterile procedure. Repeated bladder
infections are not unusual during the first few months of self-catheterization
but are reduced as the bladder begins to tolerate the presence of bacteria and
clean technique improves. Anticholinergic drugs
can be continued in patients performing intermittent self-catheterization.
D. Surgical procedures. Severe or total
incontinence requires the use of an indwelling catheter or suprapubic catheter. There seems to
be little difference in the development of infection in these two techniques,
but some patients find a suprapubic catheter
with continuous drainage into a catheter bag more convenient than the transurethral catheter, and
the choice should be offered,
when appropriate.
Augmentation
cystoplasty to increase the storage volume in a severely contracted
hyperreflexic bladder is of occasional benefit for patients who can perform
self-catheterization.
21. Intention tremor is a common sign in
multiple sclerosis and many patients develop resting tremor enhanced by action
in the later stages of the disease. These are difficult symptoms to control. Devices
to dampen tremor, such as weights applied to the wrists, are of limited
benefit. Medication is unpredictable. Propranolol
(Inderal) beginning 20 mg tid and increasing to as high as 240 mg/day
when the long-acting preparation can be used, may be effective in some cases.
Other drugs of occasional benefit include clonazepam,
primidone, and hydroxyzine. Carbonic
anhydrase inhibitors such as acetazolamide and Neptazane may help in some
cases, and isoniazid in high doses has been reported to decrease tremor, but
adverse effects are not uncommon. In many cases, a combination of drugs is the
most effective approach to this problem.
22. Unsteadiness (ataxia). No medications are available to modify ataxia. The physician must persuade the
patient who is at risk from falling to take reasonable precautions to reduce
the effects of ataxia. Light-weight
wheeled vehicles with hand brakes are useful, rather than the standard walker,
which is slow and cumbersome. Severe ataxia is
an indication for the use of an electric cart, even though the patient has
little or no weakness, and this should be encouraged at an early stage, because
improvement in ataxia is unusual.
23. Contractures.
Paraplegia and flexion with knee and hip contractures
are common manifestations of neglect and should not happen in a well
planned treatment program. Contractures can
be prevented by physical therapy, appropriate splinting, and the use of
antispasticity agents such as baclofen or
tizanidine. If contractures are
established, early surgical intervention is necessary to release joints and
restore normal limb posture. The baclofen pump
has major benefit when contractures are
the result of severe flexor spasticity.
24. Diplopia is
often temporary during an exacerbation of multiple sclerosis and should be
treated by patching one eye. The patch should be alternated over each eye
daily. When diplopia is an established
symptom, the use of prisms in eyeglasses may help. Surgical correction by an
ophthalmologist is needed occasionally.
25. Impairment of bowel control. Bowel incontinence
can be a devastating event in patients with multiple sclerosis, sufficient in
some cases to convert an outgoing, gregarious patient into a recluse. The
problem can be solved by development of the innate but dormant gastrocolic reflex. The patient is instructed
to attempt bowel movements immediately after breakfast each day. The reflex
may not function for several weeks, but eventually, it will return and bowel
evacuation becomes an automatic function in the morning. The patient is then
free of worry about incontinence for the rest of the day.
Constipation
is a common complaint. A stool softener such as docosate sodium 100 mg b.i.d.
or bulk agents such as Metamucil may suffice in mild cases. When constipation
is established, the patient should take 30 mL Milk of Magnesia, plus 2 senna
tablets (Senokot) at night, if there has been no bowel movement for 2 days.
This should result in a bowel movement after breakfast the next morning. An
alternative method is to use lactulose syrup
1 to 2 tablespoons daily. In refractory cases, bisacodyl suppository
(Dulcolax) 10 mg can be used in the morning. Failure of bowel movement after
these measures requires the periodic use of an enema.
Fecal impaction is a problem in bedridden or immobile
patients. Manual removal of fecal material followed by enemas may be necessary.
The venerable but extremely effective milk and molasses enema is recommended as
a last resort.
26. Sexual dysfunction. Sexual dysfunction is
not uncommon in both men and women with multiple sclerosis, occurring in almost
80 percent of men with advanced multiple sclerosis and in approximately 50
percent of women with similar disability.
In men,
failure to achieve erection rarely responds to oral therapy with yohimbine or hormonal replacement, with parenteral testosterone which, in reality,
should only be used for hypogonadal disorders. Penile prostheses are cumbersome and subject to infection. Intercavernous injection of alprostadil (prostaglandin E) or propiverine combined with
phentolamine increases arterial inflow into the penis, while decreasing venous
outflow. Both methods are effective, with restoration of ability to achieve
satisfactory intercourse in the majority of cases. Adverse effects include
mild pain and dizziness. The dose of alprostadil or propiverine should be
titrated to achieve a satisfactory but not prolonged erection. Adverse effects
are infrequent and include prolonged erection, priapism,
hematoma, hypotension, and fibrosis.
Another
method using transurethral suppositories
of alprostadil is an equally successful alternative therapy, with fewer adverse
effects. It is probable that sildenafil citrate
tablets will supersede most methods for improving erectile function in males.
The use of this drug is effective in most cases of erectile dysfunction with
few serious adverse effects, the most common of which are headache in 16% of patients,
flushing in 10%, and dyspepsia in 7%.
Female
sexual problems include loss of vaginal sensation or lubrication. The latter
can be treated with vaginal lubricant. Loss of vaginal sensation requires
empathy and understanding by a concerned partner who is prepared to assist in
the development of satisfactory sexual foreplay.
Loss of
vaginal sensation can be treated in some cases with a vibrator, which enhances
sensation in the vaginal area.
27. Cognitive dysfunction. About 40 percent of
patients have cognitive difficulties, which are usually mild. The main deficits
relate to retention and recall, short-term memory, attention, and delayed
processing of information. When these difficulties interfere with the ability
to function, neuropsychological testing
should be performed to measure the extent of deterioration and to exclude
depression, which is a major cause of impaired memory in multiple sclerosis. A
small percentage of patients do show significant disability with cognitive
impairment, sufficient to interfere with daily activities. These situations
can be treated with a cognitive rehabilitation program, allowing the patient to
circumvent or minimize these difficulties. Dementia, if sufficient to produce
declining ability to function independently, is rare" and often associated
with atrophy of the corpus callosum.
28. Respiratory impairment treatment should be
directed to the control of infection followed by intravenous corticosteroid therapy. The dictum—if in doubt,
don't wait, intubate and place the
patient on a ventilator—is applicable in these cases.
29. Bulbar dysfunction.
Dysarthria is not unusual in multiple
sclerosis but is rarely of sufficient magnitude to interfere with
communication. Should this happen, the services of a speech pathologist are
indicated.
Dysphagia is, however, common and often
undetected in patients with multiple sclerosis. This involves a disturbance
of both the oral and pharyngeal phase of
swallowing, and evaluation by a speech therapist, including radiological
investigations with a barium swallow and visual fluoroscopy, is indicated. The speech pathologist can then suggest
changes in diet and the use of various maneuvers to facilitate swallowing. In
advanced cases, when dysphagia is a major
problem, the patient should be fed through a percutaneous gastrostomy tube.
30. Community services. Patients with severe
multiple sclerosis who are unable to continue their employment, or who become
increasingly dependent on others, face the prospect of growing problems at home
and in the community. Most are not equipped to cope with the stress of chronic
illness and should receive social service assessment and advice whenever
possible. This results in a smoother transition from hospital to home and
better adjustment to home conditions, with improved support for the patient and
the family. To this end, identification and referral to community resources
available to multiple sclerosis patients should be implemented in all cases
with more than a minor degree of disability.
31. Simple prophylactic measures.
A. Combat infection. In many cases, a relapse
occurs after an infection. It is prudent, therefore, to treat all infections in
multiple sclerosis patients seriously and to resort to the early use of
antibiotics. This will not have any effect on viral infections but antibiotic
therapy will reduce the risk of secondary bacterial infections such as
sinusitis, bronchitis and pneumonia.
B. Avoid fatigue. Some patients note relapse
following periods of unexpected exercise. Ambulatory patients with multiple
sclerosis should avoid sudden unexpected athletic activities or prolonged exertion.
Those who wish to exercise should develop an incremental program of activity
and stop whenever they experience fatigue.
C. Emotional stress. As a group, multiple
sclerosis patients experience more emotional stress than those who are well
There are increased rates
of divorce more financial problems, and fewer opportunities for gainful
employment. Substandard care for the chronically disablec and limitations on
mobility because of ; lack of transportation or lack of propei building access
adds to the emotional dis tress of multiple
sclerosis sufferers Whether such
stress leads to exacerbatioi is debatable, but every effort should b< made
to reduce emotional distress by ap propriate treatment or referral to commu
nity agencies.
Patients
with depression will oftei benefit from the use of appropriate antide pressants
such as fluoxetine (Prozac), ser traline (Zoloft), or paroxetine
(Paxil) which can be combined with psychothet apy in appropriate cases.
D. Avoid excessive, prolonged expo sure to
sunlight. Patients with multipl sclerosis experience considerable weak ness
with the reappearance of previou symptoms if exposed to a hot environ ment, in
particular, after prolonged expo sure to sunlight. The patient should b warned
about this possibility and reas sured that the
symptoms will resolv once the
body core temperature
de creases.
Prognosis ;
The prognosis
in multiple sclerosis has im proved in the last two decades. The mean surviva
is now 20 to 25 years. This can be attributed to bette treatment and control of
infection in debilitated pa tients. The physician should be frank with the
patient and relatives in discussing the prognosis. Multiple sclerosis resembles
a chronic infectious disease in presentation and prognosis. Consequently,
multiple sclerosis may be benign, relapsing and remitting, primary or
secondary progressive, severely disabling, or fatal. In general, patients who
present with mild symptoms and who have several mild relapses tend to remain in
the mild category and do not become severely disabled. Approximately 20
percent of patients remain fully active and fully employed 10 years after the
diagnosis of multiple sclerosis and should be informed that they have a benign
form of the disease and will not be disabled by multiple sclerosis. However,
these patients must also be informed that mild exacerbations of multiple
sclerosis will continue at unpredictable times well past the fiftieth year.
More than 50 percent of patients continue to work full-time, whereas 33 percent
are paraparetic, paraplegic, or
quadriplegic, and 25 percent require intermittent or constant catheterization for bladder dysfunction.
The
prognosis of multiple sclerosis can be improved by avoiding any precipitating
factors. The patients should be advised to identify and treat infections
promptly, to avoid unusual physical or emotional stress, and to avoid prolonged
exposure to sunlight. All infections should be treated with the early use of
antibiotics. Patients with chronic multiple sclerosis who are bedridden or
confined to a wheelchair often experience slow deterioration, which is not
appreciated by patients or by relatives. The prognosis can be improved in
these cases by regular reevaluation at 6-month intervals, followed by prompt
attention to obvious areas of deterioration. Patients with urinary tract
problems should be reevaluated frequently.
Loss of function in the limbs calls for prompt reinstitution of physical
therapy and treatment with intravenous corticosteroids.
Paraparesis should not render a patient bedridden. Prescription of the
correct type of wheelchair and instruction in the proper transfer from bed to
chair permit broader contact with friends and relatives, improve morale, and
improve the long-term outlook for the patient. Pregnancy is associated with
clinical stability in most cases, but the postpartum
period carries a risk of exacerbation of multiple sclerosis by two or
three times the expected relapse rate. Patients should be told that there is
some increased risk of multiple sclerosis in the offspring if one parent has
the disease, but the actual risk is small.
Acute
Disseminated Encephalomyelitis
This
condition is an acute demyelinating disease believed to be an autoimmune
response to a systemic viral infection. The illness may begin within a week or
as long as 4 weeks after an acute viral infection such as measles, varicella,
or rubella, and rarely following mumps or influenza (postinfectious encephalomyelitis). In many cases, the infectious
component is unrecorded or may present as a mild upper respiratory tract
infection. Acute disseminated encephalomyelitis
used to be a recognized complication of vaccination against rabies or
smallpox (postvaccinal encephalomyelitis), but
this condition has largely been eliminated by the use of modern rabies vaccines
and by the eradication of smallpox worldwide. Occasional cases may be seen
following injection of tetanus antiserum or
earlier forms of rabies vaccine, which are still in use in Third World
countries.
Pathology
The brain
and spinal cord show the presence of perivascular
cuffing with lymphocytes and plasma cells and scattered areas of perivascular demyelination. The primary lesion
is believed to be a vasculopathy and discrete areas of hemorrhage are seen in
some cases. The condition has a similar pathological appearance to experimental
allergic encephalomyelitis. In some
fulminating cases, the brain is swollen and shows the presence of numerous petechial hemorrhages or hematoma formation. Microscopic changes
consist of necrosis of blood vessel walls and the passage of fibrinous exudate into the perivascular spaces. There are marked white
matter edema and infiltration of abnormal areas with inflammatory cells. The
affected blood vessels may be surrounded by areas of necrosis and
demyelination or ball or ring-like hemorrhages (acute hemorrhagic encephalomyelitis).
Clinical
Features
The decrease
in the incidence of acute disseminated encephalomyelitis
has been attributed to increased vaccination and immunization against
infectious diseases. The onset is usually abrupt, occurring within a week of
clinical evidence of infection, but the first symptoms have been reported
before any signs of infection, and as long as 2 or 3 months after infection.
The early symptoms
consist of
headache, fever, anorexia, and lethargy. The subsequent course shows
considerable variation. The illness may resemble a mild encephalitis with
complete recovery, or there may be rapid progression to stupor and coma.
Psychiatric symptoms consisting of personality changes, hallucinations, or
acute paranoia are not uncommon. Seizures, sensory or motor disturbances, or
dysfunction of bladder or bowel may occur.
Differential
Diagnosis
The
disseminated involvement of the CNS in acute disseminated encephalomyelitis produces a clinical picture
that mimics acute multiple sclerosis. The development of symptoms following
documented infections, however, suggests the diagnosis of acute disseminated encephalomyelitis.
Diagnostic Procedures
1. On lumbar puncture, the CSF is clear, with
normal, slightly elevated pressure. There is a
lymphocytic pleocytosis and red blood cells are present in some cases.
The protein and gamma globulin contents are elevated. The glucose content is
normal.
2. The EEG is abnormal in severe cerebral involvement,
with diffuse slowing in the theta and delta range.
3. The MRI scan shows the presence of numerous
areas of increased signal intensity in the white matter of the brainstem and spinal cord, and in the gray
matter of basal ganglia, thalamus, and
temporal lobe cortex.
Treatment
1. Plasmapheresis
alone or combined with high-dose corticosteroids
and cyclophosphamide may
produce a successful recovery in some cases.
2. The fulminating acute hemorrhagic form of the disease is associated
with rapid increase in ICP and requires ICP monitoring, intravenous mannitol
and high-dose barbiturate therapy.
Students’ practical Study Program.
Step I. Aim: To put
clinical diagnosis. For this purpose it is necessary:
1.
To determine clinical form of multiply
sclerosis, acute disseminated encephalomyelitis, amyotrophic
lateral sclerosis, acute myelitis by mean of scheme of differential diagnosis.
2. To
define basic clinical syndromes of locomotors, sensitive, coordinative
dysfunction, cranial innervation.
3.
Carry out differential
diagnosis between multiplex sclerosis and acute disseminated
encephalomyelitis.
4.
To formulate clinical diagnosis, for example:
a) Multiply
sclerosis, cerebral-spinal form with presence of lower spastic paralysis,
degree III, progradientive course
b)
Acute disseminated encephalomyelitis
(encephalomyelopolyradiculoneuritis) with presence of mixed tetraparesis and disturbed
sensation of radicular and polyneuritic type.
Step II. Aim: To prescribe therapy. For this it is necessary to pay attention to
pathogenetic mechanism of demyelination diseases of
nervous system, clinical forms, course of disease, presence of remission, acute
periods, est.
For
the treatment of multiply sclerosis prescribe:
-
general-clamping drags (ATPh, Cocarboxylase);
-
vitamins group B (B1, B6,
B12), ascorbinic acid (Vit C), nicotinic acid (PP);
-
hormones (prednisolone, metipred, dexamethazone,
polcortolone) in onset of disease;
-
desensibilisation drugs (dimedrol, pipolphene,
diazoline, claritin, hormones);
-
immunostimulators (T-activine,
Thymaline, Sinacten-depo, Echinacea);
-
pyrogenic drugs (pyrogenal);
-
immunodepressors (hormones,
asathyoprine);
-
myorelaxation drugs (Midocalm,
Mellictine, Bactrophene);
-
biostimulators (aloe, vitreous
body);
-
plasmapheresis.
In
causes of ADEM also it is necessary to prescribe course of antibiotics therapy
(series of penicillin, cephasolin), enzymes (desoxyribonucleinase,
ribonucleinase) and antivirus remedies such as Reapherone, Acyclovir, Zovirax,
hormones, desensibilisation medicines. Independently prescribes medicines for
therapy of amyotrophic lateral sclerosis.
Step III.
Aim: To fulfill preventive-examinative measures.
On
reason from clinical diagnosis and effect of therapy to define prognosis about
life, work, recovering, to take preventive measures, to fulfill medical labor
examination and military medical examination.
Amyotrophic
Lateral Sclerosis
Amyotrophic
lateral sclerosis is a chronic progressive disease of nervous system and is defined
pathologically as one in which there is degeneration of both upper and lower
motor neurons of brain and spinal cord. It is characterized by unfavourable
prognosis. The disease was first described by Charcot and
Joffroy in 1869.
Epidemiology
The incidence rate of Amyotrophic Lateral
Sclerosis of 2 – 5 cases per 100 000 population has been found in many
countries. As many as 60 000 - 70 000 begin_of_the_skype_highlighting ![]() 60
000 - 70 000 FREE end_of_the_skype_highlighting patients have
this disease at any one time all over the world.
60
000 - 70 000 FREE end_of_the_skype_highlighting patients have
this disease at any one time all over the world.
The disease is one of middle and late life. Only
10% of cases begin before age 40 years; 5% begin before age
The disease is found worldwide in roughly the
same prevalence. However, about 50 times that number were found on the island
of Guam.
Etiology and pathogenesis
The cause of ALS is not known. Most of
scientists consider this disease to be a polietiologic one.
The main theories:
1. Connection
of ALS with Prion Diseases. This can be proved by high content of Arginaza in
the brain of animals in experiment and in the CSF of patients with ALS. Besides
this we can find morphologic changes in the brain of such patients. They are
spongiosis and synapse degeneration. This can be explained by the deficiency of
Argininum.
2. Autoimmune
theory. There are certain changes in cell – mediated and humoral immunity.
3. Genetic
theory. The patients with familial forms of ALS have changes of gene, located
in the 21st chromosome. This gene is responsible for the production
of endogenous antioxidants. The deficiency of such antioxidants can cause
activation of free radical oxidation that leads to the death of neurons of
anterior horn.
4. Amino
acids, neuromediators, neuropeptides metabolism disorders. And those chemical
substances are responsible for the apoptosis. That’s why the patients with ALS
have low content of Mg, Mn, AL, Se in biologic fluids.
The main reasons of ALS development are:
1. Insufficiency
of spinal blood circulation.
2. Vertebrogenous pathology
3. Trauma
4. Infections
5. Demyelination
6. Metabolic
disorders
7. Tumours
(rarely)
8. Radiation


Pathomorphology
The pathologic process is localized in
·
Anterior horns of the spinal cord
·
Pyramidal tract
·
Motor neurons of IX, X, XI, XII CN’s,
sometimes - V, VII
There is degeneration of third and fifth cortex
layers of the anterior central gyrus (small and large pyramidal cells).
Besides there is demyelination of pyramidal
tracts (in the internal capsule, lateral and anterior horns of the spinal cord)
that cause the death of axial cylinder. That means that the fibres irreversibly
die.
Classification
There are three forms of the disease:
1. Classical
or sporadic ALS
2. Familial
ALS which is usually associated with
·
Dementia
·
Extrapyramidal disorders
·
Extrapyramidal and cerebellar disorders
·
Extrapyramidal and sensory disorders
·
Peripheral neuropathy
3. Complex
– ALS – Parkinson disease – Dementia
Familial ALS usually begins at the age of
45 – 48. This form can be inherited as autosomal dominant and autosomal
recessive one.
According to the localization of the process at
the beginning of the disease we can diffea such clinical forms:
1. Cerebral
2. Bulbar
3. Cervical
- thoracic
4. Lumbar
– sacral
Clinical features of the main forms of ALS and
its diagnostics
According to the
International Neurologic Community in 1990 the main criteria of ALS were
established:
·
The symptoms of lower motor neuron lesion
·
The symptoms of upper motor neuron lesion
·
Progressive course of the disease (during 6
months and more)
·
No effects from medicines and physiotherapeutic
treatment
ALS is ruled out at:
·
Sensory disorders
·
Pelvic disorders
·
Eye movements disorders
·
Autonomic disturbances
·
Also if it is not a familial form - in case of Parkinson disease and Dementia
1. Anamnesis
of the disease.
The first symptoms of the disease are weakness,
especially in extremities (hands in particular), awkwardness during the
performance of acute movements, reduced muscles of thenar and hypothenar,
fasciculations. Forerunner symptoms of ALS are crumpies (painful tension of
calf muscles). These symptoms were observed in 30% of patients 3 – 6 months
before the other signs of the disease.
2. Typical
clinical picture
·
Flaccid and spastic paralysis
·
Muscles atrophy
·
Bulbar and pseudobulbar signs
The
reflexes at the beginning are very high with wide reflexogenic zones. There are
all the groups of pathologic reflexes. But :
- Abdominal reflexes are preserved for a long time
- There are no pelvic disorders

Clinical picture of high (cerebral) form of ALS
It is observed in 1-2% of all patients with ALS.
It is characterized by spastic tetraparesis and pseudobulbar syndrome.
Sometimes at this form Extrapyramidal nervous system (the signs of Parkinson
disease) and cortex of frontal lobe (the signs of Dementia ) is involved.
Clinical picture of bulbar form of ALS
It is observed in 25 % of all patients with ALS.
At the beginning the process is localized in the medulla oblongata and causes
the lesion of IX, X, XI, XII CN’s that leads to the classic bulbar syndrome.
Then it is joined by anterior horns amyotrophy and pyramidal disorders.
Clinical picture of cervical - thoracic form of
ALS
It is observed in 50 % of all patients with ALS.
The disease begins with the weakness and reduced muscles of proximal and later
distal parts of the upper extremities. There is well – expressed hypotrophy,
increased muscle tone according to the spastic type, hyperreflexia, pathologic
signs. That means that there is a mixed paresis. In the course of the disease
the reflexes fade away, the peripheral palsy dominates, bulbar symptoms are
joined.
Clinical picture of lumbar – sacral form of ALS
It is observed in 25 % of all patients with ALS.
The first signs of the disease are connected with the peroneal muscle group
lesion. This is associated with high stretch reflexes, pathologic signs. This
form is characterized by slow progress.
Depending on the combination or domination of
some clinical signs there are:
1. Classical
type – when anterior horns and lateral columns are involved in pathologic
process in equal way.
2. Poliomyelitic
type – in 10 – 12 % of all cases the lesion of anterior horns (that means
amyotrophy) dominates.
3. Spastic
type – the conductive disorders dominate.
The stages of the disease
I. There are only
subjective signs (complains). This stage lasts for about 2 – 3 years.
II. There are the clinical
signs of the disease – focal and bulbar disorders. This stage lasts for about 6
months – 2 years.
III. The process involves neighbour regions. This
stage lasts for about 4 months – 2 years.
IV.Terminal stage. The
patients need somebody’s care. This stage lasts for about 3 months – 1 year.
The additional methods of diagnostics
ENMG:
- Regular rhythmical fasciculations
- Decreased speed of impulse conductance along motor fibres but
preserved speed of impulse conductance along sensory fibres.
This
method is very important in the diagnostics. Due to it we can:
- Localize the process
- To find out its expansion
- The degree of lower and upper motor neurons lesion
- To register fasciculations as usually it is one of the first symptoms
of the disease.
MRI
- We can diagnose atrophy and bilateral degeneration of cortico – spinal
tracts. In case of high lesion we can find the focus in the posterior crus
of the internal capsule.
PET
- One can register the low content of glucosa in the region of anterior
central gyrus. This sign by itself doesn’t have very important meaning.
CSF – examination
- Increased protein level
- Increased activity of Arginaza
Differential diagnosis should
be made with following diseases:
1. Chronic
stage of Acaridan encephalitis
2. Cervical
ischemic myelopathy, which is characterized by slow progress and often with the
termination of the process
·
Limited fasciculations, which are non –
rhythmical
·
A great attention should be paid to the X- ray –
examination, MRI, ENMG
3. Subacute
poliomyelitis which is characterized by the absence of pyramidal tract lesion
4. Spinal
amyotrophy, which is characterized by slow benign course. Pyramidal signs are
very rare at this disease
5. Syringomyelia
(anterior horns form). There are sensory and segmental trophic disorders
6. The
tumours of the cervical part of the spinal cord – intramedullar. Only MRI and
CT examination can diffe those pathologies at early stages
7. Radiculoischemia
on the lumbar level is characterized by peculiarities of development,
vertebrogenous syndrome, radicular syndrome and partial regress in case of treatment
The course of the disease and prognosis
There is progressive course of the disease.
The result of ALS in most cases is death of the patient. Sometimes the periods
of stable symptoms can be observed during 1 – 3 years.
The duration of life in such patients depending
on the clinical form of the disease:
1. Cerebral
form – 4 – 6 years.
2. Bulbar
form – 1 – 3 years
3. Cervical
– thoracic form – 6 – 8 years
4. Lumbar
– sacral form – 10 – 12, 9 – 16 years.
The main principles of treatment:
1. For
the examination and diagnosis putting the patients are sent to the neurologic
hospital
2. There
is no effective treatment for ALS
3. For
the stabilization of the process there are used :
·
Vit B (2 – 4 ml per day during 30 days)
·
Vit E 1500 mg per day
·
Biostimulators
·
Anabolics (Nerobol 5 – 10 mg twice a day during
1.5 – 2 months, Retabolil – 1 ml (50 mg) once a week)
·
As pathogenetic treatment we prescribe Delargil
2 mg twice a day i/v during 10 days, then i/m up to 1 month.
·
Tireotropinum i/v by drops 500 mg
·
Riluzan 100 mg per kg during 12 months.
·
At increased tonus myorelaxants are used.
·
Dipheninum is prescribed at crampy
·
Antidepressive medicines
·
Electro – therapy is excluded
·
The patients in the third and fourth stage need
somebody’s care. Sometimes fixative equipments are used. At bulbar disorders
the patients get food in a special way.
Motor Neuron Disease (Amyotrophic Lateral Sclerosis)
Definition Motor neuron
disease (amyotrophic lateral
sclerosis—ALS) is a chronic disease characterized by progressive degeneration
of motor neurons of the anterior horn of the spinal cord, motor nuclei in the brainstem, and neurons in the motor area of the
posterior aspect of the frontal lobe.
Etiology and
Pathology
The etiology is unknown. The disease is familial in 10 percent of cases,
when it is inherited as an autosomal dominant
or autosomal recessive trait. Dominant
familial ALS has a penetrance of less
than 100 percent by age 70 years and may be lower than 50 percent by
age 63.85 This is the most common form of familial ALS, with the responsible
gene located on chromosome 21, whereas some families with recessive ALS show
linkage to markers on chromosome 2 or 21. The mutations are associated with an
abnormality in the superoxide dis-mutases,
which are a group of isoenzymes that catalyze
the conversion of superoxide free radical
ions to hydrogen peroxide and oxygen. Failure of this reaction permits free
radicals, which are highly toxic, to accumulate in motor neurons, producing
free radical toxicity and accelerated
cell death.
Some 90
percent of patients with ALS are without evidence of chromosomal abnormality
with superoxide dismutases mutations
present in a small percentage. In the majority, however, the disease may be
the result of exotoxicity. This process postulates overactivity of the glutamate system, in which the normal amino acid is converted into an exotoxin which produces glutamate overstimulation of the five subtypes of the
glutamate receptors on the neuronal membrane,
resulting in increased calcium entry into neurons, overproduction of free
radicals, and premature cell death.
Evidence for
an autoimmune mechanism producing neuronal loss
or the role of positive viral agents such as the poliovirus are unproven.
Failure of axonal transport with
accumulation of neurofilaments, which
are known to be increased in ALS, could indicate a failure of axonal transport,
leading to dendritic atrophy and death of the perikaryon.
Pathological
changes consist of loss of motor neurons in the spinal cord, brainstem, and cerebral cortex. Surviving
neurons are atrophic with abnormalities
of dendritic pattern on Golgi staining and show reduced levels of RNA.
Inclusion bodies, identified as hyalin inclusions, Lewy bodies, basophilic inclusions, and Bunani bodies, occur
in motor neurons in familial and nonfamilial ALS. A filamentous neuronal inclusion body has been identified in
nonfamilial ALS in both motor and nonmotor neurons in < the brain,
suggesting that ALS should be considered a ' multisystem disease. '
Clinical
Features Amyotrophic lateral sclera- | sis is a rare
disease with a prevalence of approxi- j mately 2 per 100,000 and a
male-to-female ratio of i 1.1:1. The median age of onset is 66 years. ]
The
fundamental disease process in ALS involves loss of motor neurons in the
cerebral cortex, the motor nuclei of the cranial nerves, and the anterior horn
cells of the spinal cord. Consequently, the classical division of ALS into
those with bulbar onset (progressive bulbar palsy) and those with involvement of the
spinal cord motor neurons (progressive muscular atrophy) may well be artificial
because careful neurological examination will reveal evidence of lower motor
neuron involvement in patients with predominantly bulbar symptoms, whereas those with < spinal motor neuron
signs of wasting and fasciculations usually
present with increased tendon reflexes and extensor plantar responses,
indicating that the pathological process includes the corticospinal tracts. It is probable that ALS consists of a
spectrum of disease extending from primary lateral sclerosis to spinal
muscular atrophy and that ultimately, if the patient survives, signs of both
upper and lower motor neuron disease will be apparent.
The disease
often begins with progressive loss of motor neurons in the anterior horns of
the spinal cord. When the neurons of the cervical cord are involved, there will
be progressive weakness, wasting, and fasciculations involving the small
muscles of the hands. However, the loss may occur at any site in the spinal
cord. The sensory examination is normal. Initially, there may be no clinical
evidence of corticospinal tract or bulbar involvement, which will inevitably
develop later as the disease progresses.
Alternatively,
the disease may begin with involvement of the motor neurons of the cranial
nerve nuclei. Under these circumstances, there is progressive weakness and
wasting involving the pharyngeal musculature,
the tongue, and the facial muscles. The patient develops progressive dysarthria and dysphagia.
Fasciculations of the tongue are usually prominent and can be seen when
the tongue is examined, lying quietly on the floor of the mouth. The oculomotor
nuclei are usually not involved.
Signs
indicating spinal motor neuron disease with wasting, fasciculations, the
development of corticospinal tract
dysfunction, increased tendon reflexes and extensor plantar responses,
ultimately occur in patients who survive for more than a few months.
A combined
form of upper motor neuron disease and anterior horn cell involvement, plus corticospinal tract dysfunction, is the most
common form of ALS. The patient complains of increasing weakness and
examination reveals atrophy, fasciculations, and wasting of the muscles in both
upper and lower limbs, associated with increased reflexes and extensor plantar
responses. Eventually the brainstem nuclei
are involved, resulting in dysphagia,
dysarthria, and facial weakness. There are no sensory changes.
Diagnostic
Procedures
1. Electromyography
(EMG) as an extension of the clinical examination, is the single most
useful test in evaluating disease of the lower motor neurons. The
EMG will demonstrate abnormalities in upper and lower extremities and thoracic
paraspinal muscles and the presence of normal nerve conduction velocities.
2. Muscle biopsy shows the typical appearance of
denervation atrophy with atrophic fascicles coexisting with normal
fascicles.
3. Muscle enzymes such as creatinine phosphokinase (CK) may be elevated
in rapidly progressive cases.
4. The CSF is normal.
5. There are no MRI, CT, or myelographic
abnormalities.
Differential
Diagnosis
1. With mass lesions impinging on the spinal
cord (e.g., tumors, spondylosis, abscess),
the weakness, wasting, and fasciculations are confined to one or more myotomes, and there is appropriate sensory loss
in the affected dermatomes.
2. Multifocal motor
neuropathy with conduction block can be differentiated by EMG
and the presence of GM: antibodies in this disease.
3. Combined cervical myeloradiculopathy and lumbosacral
radiculopathy with both upper and lower motor neuron signs can be
excluded if brainstem cervical, thoracic,
and lumbosacral areas are included in
the EMG evaluation.
4. Benign fasciculations, myokymia. Benign fasciculations are not at all
uncommon and are often diffuse. There are no other signs of neurological involvement,
and the general physical examination and neurological examination are perfectly
normal. Follow-up examinations remain normal.
5. Chronic inflammation of the meninges or spinal cord (e.g. adhesive
arachnoiditis). In this condition, the CSF is abnormal. The MRI and CT scans
with myelography may be abnormal.
Treatment
There is no effective treatment for this disease and the course is one of
progressive deterioration, with a mean survival of 3 years. Immunosuppressant and intravenous immunoglobulin therapies are not effective. Selegiline does not modify the progression of
ALS. Ceftriaxone is without benefit.
Lamotrigine and gabapentin, which inhibit glutamate release, do not alter the
course of this disease. Neither dextromethorphan,
physostigmine, branch-chain amino
acids, 3,4-aminopyridine, nor acetylcysteine
has had a significant effect on ALS. Recombinant
human insulin-like growth factor-1 is reported to slow the progression of
ALS and deterioration in the quality of life.
Riluzole, a
glutamate antagonist, is reported to increase survival in ALS and appears to be
most effective in patients with disease of bulbar
onset. The drug is taken by mouth, 50 mg ql2h, 1 h before or 2 h after a
meal. Adverse effects include nausea, weight loss, and headache.
There should
be close support by physicians, nurses, social workers, and psychologists to
help the patient face the ravages of this disease. The family should be
informed of the course complications and prognosis of the disease. Community
resources should be obtained through the help of a social worker because the
family will require increasing support as the patient becomes increasingly dependent.
When dysphagia prevents normal feeding,
feedings should be continued through a plastic naso-gastrostomy or jejunostomy
tube, using a pureed diet and ultimately
the use of a percutaneous gastronomy (PEG) tube. The weakness and wasting often
produce painful subluxation of the scapulohumeral joints, and the arms should be
supported by hemiplegic slings. Adequate analgesia for pain control is
essential. Aggressive symptomatic treatment should focus on the goal of
maintaining the patient's functional independence as long as possible. This
requires an ongoing program of physical rehabilitation, including physical and
occupational therapies, the use of assistive devices,
the provision of home equipment, and nutritional, communication, respiratory,
and psychological support.
When
respiratory failure occurs, the family should be assured that the patient will
pass quietly into a carbon dioxide narcosis, followed by coma and death, and
should be urged to avoid the use of a mechanical respirator in most cases.
However, a number of patients develop respiratory failure at an early stage,
when there is still adequate function in the upper limbs. These individuals
may lead a useful life of
2 or more
years, in some cases, with portable mechanical ventilator support. Death
occurs from pulmonary infection as the patient becomes immobilized by muscle
weakness.
The bulbar musculature tends to lose function at a
slower rate than the muscles of the upper limbs, but bulbar involvement is more critical because of the dangers of
aspiration pneumonia.
Prognosis
The course
is one of steady progression until death. Life expectancy of patients with dysphagia is less than that of patients with
limb weakness, presumably because of the higher risk of aspiration pneumonia in
the dysphagia group. Occasionally, the
process seems to be arrested for a period of several months before continuing
a downhill course. Mean survival in the familial form of ALS is 3 years, with mean survival of 4 years in
sporadic cases of ALS.
Primary
Lateral Sclerosis
Definition This is a slowly progressive disorder of nonfamilial form, in which there
is a gradual development of spastic paraparesis over
a period of many years.
Etiology and
Pathology
The etiology
is unknown. Pathological changes consist of frontal lobe atrophy with neuronal loss and corresponding reduction in subcortical white matter, involving the
precentral gyrus, the premotor areas, and
the supplementary motor areas, with degeneration of the corticospinal and corticobulbar axons.
The pathological changes suggest that there may be an overlap with ALS
and frontotemporal dementia.
Clinical
Features
The
condition presents in adults with a gradual development of spasticity in the lower limbs, producing
progressive impairment of gait and eventual requirement of a cane or walker.
The upper limbs are spared but show hyperreflexia. The tendon reflexes are
increased in the lower limbs, and bilateral extensor plantar responses are
present. Spastic dysarthria and mild dysphagia are late developments. There is no
evidence of amyotrophy, fasciculations,
optic atrophy, hearing loss, sensory change, or pes
cavus. There are no sphincter problems, no urgency of micturition, and
absence of any evidence of a segmental lesion.
The clinical
course is slow, with a median survival of several years. Some patients show
evidence of muscle wasting late in the course of the disease, suggesting that
primary lateral sclerosis is a forme fruste of ALS or frontotemporal dementia.
Diagnostic
Procedures
1. An MRI or CT scan should be obtained to
exclude malformations at the craniocervical junction, cervical spondylosis, spinal canal or canal tumors, and
multiple sclerosis.
2. The CSF is normal.
3. A SPECT or PET scan shows bilateral posterior
frontal lobe hypoperfusion/hypometabolism.
4. Neuropsychological
testing may reveal minor cognitive deficits that escape detection in a
brief mental status evaluation.
Treatment
Oral baclofen or a
baclofen pump may be useful in reducing spasticity.
Adequate evalu ation will avoid unnecessary surgery
for higher cervical cord compression.
TRANSVERSE MYELITIS (MYELOPATHY)
Definition
Transverse
myelitis is a syndrome characterized by
acute spinal cord dysfunction involving both halves of the cord in transverse
section.
Etiology and
Pathology
The various
etiologies of transverse myelitis are outlined in Table 12-2. The condition
may be a periinfectious or postinfectious process
and has been associated with many viral infections, including poliovirus,
echovirus, and Coxsackieviruses.12 In
some patients, there is direct viral involvement of the spinal cord, whereas
others present with a postinfectious process,
probably the result of inflammation, edema, or an autoimmune response to viral
infection. Transverse myelitis has been associated with measles, varicella,
mumps, rabies, typhoid, systemic lupus erythematosus,
and a reaction to sulfonamides. Transverse
myelitis has also followed vaccination and immunization against rubella,
diphtheria, and poliomyelitis. Vascular occlusion with softening of the cord
may occur in syphilitic arteritis and in
the arteritis associated with collagen
vascular diseases. Arterial occlusion due to a dissecting aneurysm of the aorta or after surgical
resection of an aortic aneurysm are other
possible causes. Acute transverse myelitis in heroin addicts is probably due to
focal arteritis. Transverse myelitis may
occur as an acute demyelinating process in multiple sclerosis and a severe
acute myelitis as a remote effect of carcinoma. The increasing survival of
patients with treated neoplasms has led to the recognition of increasing
numbers of cases of acute transverse myelitis secondary to radiation therapy.
Table 12-2
Etiologies of transverse myelitis
1.
Congenital—vascular malformation
2.
Infectious—viral infection
3.
Autoimmune—peri-infectious, postinfectious, or vaccinial myelitis
4.
Multiple sclerosis
5.
Neoplastic—paraneoplastic necrosis
6. Toxic—secondary to heroin injections
7. Vascular—vascular insufficiency—arteritis, dissecting aneurysm, aorta resection, aortic aneurysm
8. Degenerative—irradiation
9. Idiopathic
Sudden
bleeding from vascular malformations or capillary telangiectasia may produce a similar picture.
The
pathological changes vary. In most cases, there is necrosis of the cord, often
involving several segments. The necrosis is maximal in the center of the cord
and even in severe cases, there is always a thin rim of surviving tissue at the
periphery. The posterior nerve roots and posterior root ganglia are occasionally
involved in the process. In cases of vascular etiology, there is infarction of
the spinal cord.
Clinical
Features
A history of
recent acute illness suggesting a viral or bacterial infection may be obtained
in about one-third of the patients. The most common complaint is a history of
upper respiratory tract infection or a flu-like illness. Transverse myelitis is
occasionally preceded by a gastrointestinal illness. Patients with transverse
myelitis occurring as a paraneoplastic condition
have a history of pre-existing neoplasia that
is frequently metastatic. Radiation
transverse myelitis occurs several months to a year after radiation therapy.
Transverse
myelitis presents with symptoms and signs indicating involvement of gray matter
and the corticospinal and spinothalamic tracts.
1. Paresthesias,
which are often described as numbness, tingling, or pins and needles,
usually begin in the toes or the feet and extend up the lower limbs into the
trunk, with eventual involvement of the upper limbs in cervical transverse
myelitis.
2. Pain is of sudden onset, usually severe and
corresponding to the level of the cord involvement. Consequently, the pain is
often in the intrascapular region.
3. Progressive lower limb weakness is noted,
often presenting with "stumbling or weakness of one leg."
4. Urinary retention occasionally occurs as
the initial complaint and is usually followed by lower limb weakness in a short
period of time.
The course
of transverse myelitis may vary:
1. A smooth progressive course often begins
with involvement of the lower limbs followed by ascending paresthesias and weakness over a period of 2
weeks, at which time the condition stabilizes with paraplegia or quadriplegia
and a well-defined sensory level.
2. In subacute progression
the symptoms appear intermittently over a 10-day to 4-week period. During this
time, the illness may appear to stabilize, only to be followed by the
appearance of new symptoms after several days of apparent stabilization.
3. Progressive limb weakness often presents
with stumbling or asymmetrical weakness of a lower limb or with upper limb paresthesias when there is cervical cord
involvement.
4. An
acute catastrophic illness
with all symptoms develops
within 12 h and sometimes in less than an hour after onset. This type of
presentation is usually preceded by marked back pain. Recovery is slow and
incomplete.
Examination of the patient reveals a
flaccid paresis or paralysis with depressed or absent tendon reflexes
suggesting involvement of anterior horn cells. There is bilateral symmetrical
sensory loss extending to the upper level of cord involvement. This site
commonly lies between T6 and T12, but there may be extension of the cervical
cord in some cases. The sensory loss is usually total, involving touch,
pinprick, vibration, and proprioception, or
the loss may be dissociated, with loss of pinprick and preservation of posterior
column functions. Bladder distention occurs
early in the illness in the majority of patients and is followed by overflow
incontinence. Fecal incontinence is less frequently encountered.
Diagnostic
Procedures
1. The T2-weighted MRI scan shows areas of increased
signal intensity involving gray matter and surrounding white matter.
2. Electromyography
performed after 2 weeks will demonstrate motor axonopathy in keeping with anterior horn cell involvement.
3. Lumbar puncture. The CSF is abnormal with a
pleocytosis in about 50 percent of cases, which varies from a mild increase in
leukocytes to a white count as high as 300 cells per cubic millimeter. The
cells are predominantly monocytic. Protein
content is elevated in about 40 percent of cases.
Differential
Diagnosis
See Table
12-1.
Treatment
The patient
should be placed on bed rest and turned every 2 h to prevent the development of
decubiti and also to promote drainage of the dependent portions of the lungs.
The bladder should be catheterized and the urine cultured periodically to
detect any infection. Appropriate antibiotics are inducted if infection occurs.
The bowels should be moved once every 2 days and a diet with adequate bulk content
given as soon as possible to prevent fecal impaction.
Physical therapy with passive movements of all joints should be
performed twice a day. Application of foam rubber splints to the lower limbs
helps to prevent contraction deformities. Patients with involvement of the
upper limbs require adequate splinting of the forearms,, wrists, and hands.
As recovery
occurs, efforts should be made to I remove the bladder catheter as soon as
possible and urological evaluation should be obtained to determine bladder
function. Stimulants such as bethanechol chloride, a parasympathetic stimulant, 10 to 50 mg three or four times a day
may help to initiate micturition and empty the bladder in patients with
persistent | bladder paralysis.
Spasticity involving the lower limbs can
be re-1 lieved by baclofen (Lioresal) 10
mg q6h, increasing slowly to as high as 100 to 120 mg/day. Severe spasticity will require the use of a baclofen pump with intrathecal injection of baclofen.
Physical
therapy, beginning with passive therapy in the acute phase, should be changed
gradually to a more active program as the patient recovers.
Prognosis
The majority
of patients with transverse myelitis show some degree of recovery, which varies
from complete to minimal. More than 50 percent of patients with the idiopathic form of the disease regain the
ability to walk within a year, although a number will be dependent on
orthopedic appliances. Twenty-five percent of patients show poor recovery and
the majority of these give a history of severe back pain preceding a rapid
catastrophic onset of transverse myelitis.