MANAGEMENT OF PATIENTS WITH AUTOIMMUNE CRISIS. MANAGEMENT OF PATIENTS WITH ACUTE GOUTY ATTACK
Gout
Gout is an
inflammatory arthritis characterized by self-limiting but excruciatingly painful acute attacks. These are a consequence of monosodium urate (MSU) crystal
deposition within articular or
periarticular tissue. After years of acute intermittent
gout, chronic tophaceous gout can develop. Tophi,
nodular masses of uric acid (UA) crystals, can form
anywhere but most commonl affect finger tips or hands.
Recent advances in understanding of intracellular events
have occurred along with new treatment development.

This illustration is by the
caricaturist, Jame Gillray (1757-1815). The image conveys a feeling of intense
pain brought on by gout
History
Hippocrates and many other
physicians of the ancient world were acquainted with the symptoms and signs of gout, but it was Paul of Aegina, who lived a
thousand years after Hippocrates, who apparently was the first physician to
comment that emotional factors, among others, could set off an attack of acute
gouty arthritis. Many since have noted that emotional stress and dietary
indiscretion seem to be the two most common proximal antecedents of the acute
episode. Any mention of the history of
gout must include the remarkable fact that some of the most notable personages
in history, including artists, philosophers, poets, scientists, physicians,
soldiers, rulers and statesmen, have supposedly been afflicted by gout (A
partial listing includes King Priam, Oedipus, Ulysses, Alexander the Great,
Charlemagne, Michelangelo, the Medici Family, Henry VII and VIII, Cardinal
Wolsey, Lord Burghley, Erasmus, John Calvin, Martin Luther,Oliver
Cromwell, James 1, John Milton, Louis XIV, Sir Isaac
Newton, William Harvey, Samuel Johnson, Sir Horace VValpole, Thomas Sydenham, William Pitt the Elder and
the Younger, Lord Chesterfield, Edward Gibbon, Lord Beaverbrook, Benjamin
Franklin, Alexander Hamilton, and Theodore Koosevelt.) This fact was commented
upon in 1778 by Cullen, himself a sufferer from gout, who
noted that the disease "more frequently attacks the wise than the
foolish." This impression continues to linger wilh many students of gout.
On the other hand, the fact that they had gout islikely to be recorded by
history only for the great and famous; obviously, many whom history does not
mention were also afflicted by it. Surely, correlations between fame,
creativity, genius, or notoriety and, for instance, mental illness,
tuberculosis, malignancy or heart disease also could be and have been made.
Nevertheless, later in this paper we shall review data which do suggest a
correlation between hyperuricemia and certain achievementrelated variables.
Hence, Cullen's original observation may have been
indeed valid and not
based on investigator bias or sampling
error.
Epidemiology
Gouty
arthritis is predominantly a problem of post-pubertal males and is seldom seen
in women before the menopause. It is the most common cause of inflammatory
joint disease in men over 40 years old. In a typical UK general
practice of 2000 patients, there may be 17 men and 3 women with gouty arthritis
and 10 times that number with asymptomatic hyperuricaemia. Serum uric acid
concentrations are distributed in the community as a continuous variable and
are determined by a number of demographic factors, of which age, sex, body bulk
and genetic constitution are the most important. Serum uric acid levels are
higher in urban than in rural communities and are positively correlated with
intelligence, social class, weight, haemoglobin, serum proteins and a high
protein diet.
Etiology
More
than 99% of primary gout cases are referred to as idiopathic, meaning that the
cause of the hyperuricemia cannot be determined. Primary gout is most likely
the result of a combination of genetic, hormonal, and dietary factors.
Secondary gout is caused by Purines can be generated by the body itself (via
the breakdown of cells in normal cellular turnover) or can be ingested in purine-rich
foods (e.g. seafood, beer). Most people with gout, however, do not produce more
than the normal amount of uric acid. Instead, most people with gout tend to be
underexcretors. The kidney is responsible for about one third of uric acid
excretion, with the gut responsible for the rest. It may be possible that
defects in the kidney that may be genetically determined are responsible for
the predisposition of individuals for developing gout.
Secondary gout and
hyperuricemia can be promoted by:
►Eating too many food
rich of purine such as: shellfish, organ meats: liver, kidney and brain.
►Dried beans, peas and
anchovies are also rich in purines.
►Drinking too much
alcohol interferes with the body's ability to get rid of extra uric acid.
►Exposure to high levels
of lead.
►Being overweight.
►Polycythaemia
►Certain diseases lead to
excessive production of uric acid in the body e.g. of these diseases include
Leukemia, diabetes, lymphomas, and hemoglobin disorders.
►Certain drugs: which
interferes the ability of kidney to excrete uric acid, such as thiazide
diuretics, low-dose aspirin, and tuberculosis medications (pyrazinamide and
ethambutol) can also cause gout.Patients who are on cyclosporine (medication
used in transplantation ot prevent the organs rejection).
The following factors increase
your risk for gout:
- Advancing age
- Male gender
- Family history
of the condition
- Obesity
- Use of certain
drugs, including diuretics ("water pills"), low-dose aspirin,
cyclosporine, or levodopa
- Binge drinking
- Lead toxicity
- Organ transplants
- Thyroid problems
- Other serious
illness
Each risk factor is discussed
in more detail below.
Age
Middle-Aged
Adults. Gout
usually occurs in middle-aged men, peaking in the mid-40s. It is most often
associated in this age group with obesity, high blood pressure, unhealthy
cholesterol levels, and heavy alcohol use.
Elderly. Gout can also develop in
older people, when it occurs equally in men and women. In this group, gout is
most often associated with kidney problems and the use of diuretics. It is less
often associated with alcohol use.
Children. Except for rare inherited
genetic disorders that cause hyperuricemia, gout in children is rare.
Gender
Men. Men are significantly at
higher risk for gout. In males, uric acid levels rise substantially at puberty.
In about 5 - 8% of American men, levels exceed 7 mg/dL (indicating
hyperuricemia). However, gout typically strikes after 20 - 40 years of
persistent hyperuricemia, so men who develop it usually experience their first
attack between the ages of 30 and 50.
Women. Before menopause, women have
a significantly lower risk for gout than men, possibly because of the actions
of estrogen. This female hormone appears to facilitate uric acid excretion by
the kidneys. (Only about 15% of female gout cases occur before menopause.)
After menopause the risk increases in women. At age 60 the incidence is equal
in men and women, and after 80, gout occurs more often in women.
Family History
A
family history of gout is present in close to 20% of patients with this
condition. Three genetic locations have been associated with the body's uric
acid handling and gout. Some people with a family history of gout have a
defective protein (enzyme) that interferes with the way the body breaks down
purines.
Obesity
Researchers
report a clear link between body weight and uric acid levels. In one Japanese
study, overweight people had two to more than three times the rate of
hyperuricemia as those who maintained a healthy weight. Children who are obese
may have a higher risk for gout in adulthood.
Medications
Thiazide
diuretics are "water pills" used to control hypertension. The drugs
are strongly linked to the development of gout. A large percentage of patients
who develop gout at an older age report the use of diuretics.
Several other medications can
increase uric acid levels and raise your risk for gout. These include:
- Aspirin -- low
doses of aspirin reduce uric acid excretion and increase the chance for
hyperuricemia. This may be a problem for older people who take baby
aspirin (81 mg) to protect against heart disease.
- Niacin (used to
treat cholesterol problems)
- Pyrazinamide
(used to treat tuberculosis)
Alcohol
Drinking
excessive amounts of alcohol can raise your risk of gout. Beer is the kind of
alcohol most strongly linked with gout, followed by spirits. Moderate wine
consumption does not appear to increase the risk of developing gout.
Alcohol use is highly
associated with gout in younger adults. Binge drinking particularly increases
uric acid levels. Alcohol appears to play less of a role among elderly
patients, especially among women with gout.
Alcohol increases uric acid
levels in the following three ways:
- Providing an
additional dietary source of purines (the compounds from which uric acid
is formed)
- Intensifying
the body's production of uric acid
- Interfering
with the kidneys' ability to excrete uric acid
Pathogenesis
Uric acid ------►Crystals------►Crystals
deposits in joint------►Joint inflammation
Biologically
significant hyperuricemia occurs when serum urate levels exceed solubility
(~6.8 mg/dL). Hyperuricemia is a common serum
abnormality that does not always progress to gout. Humans generate about 250 to
750 mg of uric acid per day. The uric acid comes from dietary purines and the
breakdown of dying tissues. The exact cause of gout is not yet known, although
it may be linked to a genetic defect in purine metabolism. Uric acid, the most
insoluble of the purine substances, is a trioxypurine containing three oxygen
groups. The pathogenesis of gout starts with the crystallization of urate
within the joint, bursa, or tendon sheath, which leads to inflammation as a
result of phagocytosis of monosodium urate crystals; the disease is usually
associated with an elevated concentration of uric acid in the blood.
Specifically, uric acid is a breakdown product of the purines adenine, guanine,
hypoxanthine, and xanthine. Adenine and guanine are found in both DNA and RNA.
Hypoxanthine and xanthine are not incorporated into the nucleic acids as they
are being synthesized, but they are important intermediates in the synthesis
and degradation of the purine nucleotides. Both undissociated uric acid and
monosodium salt, which is the primary form found in the blood, are only
sparingly soluble. The amount of urate in the body depends on the balance
between dietary intake, synthesis, and excretion. In people with primary gout,
defects in purine metabolism lead to hyperuricemia, or high levels of uric acid
in the blood. This can be caused by increased production of uric acid, abnormal
retention of uric acid, or both. Urate in the blood can accumulate either
through an overproduction or an underexcretion of uric acid. Hyperuricemia results from the overproduction of urate found in
10% of gout patients and from underexcretion of urate found in the remaining
90%. The majority of patients with endogenous overproduction of urate have the
condition as a result of salvaged purines arising from increased cell turnover
in proliferation and inflammatory disorders, from pharmacologic intervention
resulting in increased urate production, and from tissue hypoxia. The renal
mechanism for handling urate is one of glomerular filtration followed by
partial tubular reabsorption. The final fractional excretion of uric acid is
about 20% of what was originally filtered. Uric acid levels independently
predict renal failure in patients with preexisting renal disease. Hyperuricemia
causes interstitial and glomerular changes that are independent of the presence
of crystal, and the changes very much resemble what hypertensive changes would
look like chronically. In addition, serum hyperuricemia is epidemiologically
linked to hypertension and seems to be an independent factor for the
development of hypertension. Finally, hyperuricemia is defined as a serum uric
acid level greater than 6.8 mg/dL. Serum uric acid can be normal, especially
during the gout attack. The target goal for uric acid treatment is to achieve a
level less than 6.0 mg/dL.
Classification
Etiopathogenetic
1. primary
2. secondary
Сlinical forms:
- typical acute attack of gouty
arthritis;
- pseudophlegmonous form;
- rheumatoid form;
- subacute form;
- psoriatic;
- abortive;
- extra-articular form
In the clinical development of gout has four stages:
• Asymptomatic
• Acute
• Intercritical;
• Chronic
According to the character of time joint damage:
• Acute arthritis - an inflammation
of the joints produration of no more than 3 weeks;
• Intercritial - from 3 to 12 weeks;
• Chronic - more than 12 weeks.
Periods:
- preclinical,
- intermittent (acute recurrent),
- chronic.
Variants of the course:
- mild,
- moderately,
- severe.

Phase:
1. exacerbation
2. remission
Radiographic stage of joint
damage:
1. I - large cysts (tophi) in the
subchondral bone, and in the deeper layers, sometimes sealing of soft tissue;
2. II - large cyst near the joints
and minor erosion of the articular surfaces permanent seal the periarticular
soft tissues, sometimes with calcifications;
3. III - large erosion by at least
one third of the articular surface, osteolysis the pineal gland, a significant soft tissue seal with the
deposition of lime.\
Peripheral tophi:
1. present
2. absent
The degree of functional
insufficiency:
1. 0 - function is maintained;
2. I - kept a professional
capacity;
3. II - lost a professional
capacity;
4. III - lost the capacity for
self-care.
Type nephropathy:
1. Urolithiasis.
3. Glomerulonephritis.
4. Arteriolonefroskleroz.
Clinical
features
Stages of gout. Gout has four
distinct stages:
1st stage-Asymptomatic:
Purine
is a chemical compound that is present in all of the cells of the body. Extra
purine is secreted out of the body in the urine in form of uric acid.
At times, there may be
abnormally high levels of uric acid in blood, this condition is called
"Hyperuricemia". Plasma uric acid level increases due to extra purine
secreted out in the urine in form of uric acid, but there are no symptoms. This
condition is called "Hyperuricemia".
2nd stage-Acute:
When
there is a lot of uric acid, it begins to form crystals and deposits under the
skin, forming a lump that can sometimes be felt on the outside of the body. The
first attack of gout marks the second, mild attacks usually go away quickly,
whereas severe attacks can last days or even weeks. The immune system, the
body's defense against sickness, realizes that the crystals should not be there
and starts attacking them. This is what cause joint pain, tenderness which can
be intense so that even a blanket touching the skin over the affected joint can
be unbearable.
The
metatarsophalangeal joint of a great toe is the site of the first attack of
acute gouty arthritis in 70% of patients; the ankle, the knee, the small joints
of the feet and hands, and the wrist and elbow follow in decreasing order of
frequency.
The
onset may be insidious or explosively sudden. Оften waking the patient
from sleep. The affected joint is hot, red and swollen, with shiny overlying
skin and dilated veins;it is excruciatingly painful and tender. Very acute
attacks may be accompanied by fever, leucocytosis and a raised ESRI and are
occasionally preceded by prodromal symptoms such as anorexia, nausea or a
change in mood. If untreated, the attack lasts for days or weeks but it eventually
subsides spontaneously. Resolution of the acute attack may be accompanied by
local pruritus and desquamation of the overlying skin.Some patients have only a
single attack, or suffer another only after an interval of many months or
years. More often there is a tendency to have recurrent attacks.

These
increase in frequency and duration so that eventually one attack may merge into
another and the patient remains in a prolonged state of subacute gout. Acute
attacks are occasionally polyarticular, and tenosynovitis, bursitis or
cellulitis may be the presenting feature.
Acute
attacks may be precipitated by sudden rises in serum urate following dietary
excess, alcohol, severe dietary restriction or diuretic drugs, or by sudden
falls following initiation of therapy with allopurinol or uricosuric drugs.
Acute attacks may also be provoked by trauma, unusual physical exercise,
surgery or severe systemic illness.


http://www.myfootshop.com/detail.asp?condition=gout

http://health-fts.blogspot.com/2012/03/gout.html
Similar to the previous image,
inflammation of the skin caused by gout is characterised by swelling and a smooth appearance to the skin.
The classic picture is:
►Excruciating and sudden
pain
►Stiffness in the joint
►Low-grade fever may also
be present
►Warmness
►Redness
►Swelling
The
patient usually suffers from two sources of pain:
1-The crystals inside the joint
cause intense pain whenever the affected area is moved.
2-The inflammation of the
tissues around the joint also causes the skin to be swollen, tender and sore if
it is even slightly touched. For example, a blanket draping over the affected
area could cause extreme pain.
Gout usually attacks one joint
at a time, while other arthritic conditions, such as systemic lupus and
rheumatoid arthritis, usually attack multiple joints simultaneously.
Uric acid crystals can deposit
in tiny fluid-filled sacs (bursae) around the joints. These urate crystals can
incite inflammation in the bursae leading to pain and swelling around the
joints, a condition called bursitis. In rare instances, gout leads to a more
chronic type of joint inflammation which mimics rheumatoid arthritis.
Gout usually attacks the big toe (approximately 75% of first attacks), however it can also affect other joints such as the ankle, heel, instep, knee, wrist, elbow, fingers, and spine. In some cases the condition may appear in the joints of the small toes which have become immobile due to impact injury earlier in life, causing poor blood circulation that leads to gout.
Acute gouty attacks occur in
much the same manner. Most acute gouty attacks occur in the late hours of the
night. As we sleep, our bodies tend to focus on the primary metabolic functions
such as digestion, breathing, etc. The extremities, such as the feet tend to
cool as a result of this 'lack of attention'. As they cool, and if the
dissolved amount of uric acid is high enough, the result is the change of uric
acid from a liquid to a crystal. The hallmark symptoms of gout is the acute
onset, usually at night with severe pain.
3rd stage-Intercritical:
After
the initial attack, the person enters the intercritical stage or symptom-free
interval that may last months or even years. Most gout patients have their
second attack within 6 months to 2 years from their initial episode.
4th stage-Chronic:
In the
last or chronic stage, gout attacks become frequent and become polyarticular
(affecting multiple joints at one time). Large tophi can also be found in many
joints. In advanced cases of chronic gout, the extra uric acid may also
deposits in the kidney leading to kidney stones and hypertension.
First
attacks of gouty arthritis are seldom associated with residual disability but
recurrent acute attacks are followed by progressive cartilage and bone erosion
in association with deposition of tophi and secondary degenerative changes.
Severe functional impairment and gross joint deformities may occur in chronic
tophaceous gout.
Tophi are deposits of monosodium urate crystals in soft tissue that may occur in the helix of the ear, over
olecranon processes, and over interphalangeal joints. Tophi can occur over
osteoarthritic Heberden's or Bouchard's nodes in the distal and proximal
interphalangeal joints, especially in older women. Tophus formation is related
to serum uric acid and to local factors. Tophi seldom develop in individuals
with asymptomatic hyperuricaemia; however, they may develop rapidly in the feet
or hands in post-menopausal women with heart failure and renal insufficiency
who develop acute or subacute gouty arthritis following prolonged diuretic
administration. Tophaceous gout may lead to significant morbidity and, if
untreated, can cause joint erosion and destruction. Occasionally, polyarticular
tophaceous gout presents as subcutaneous nodules that can mimic rheumatoid
arthritis. In this case, the presence of monosodium urate crystals in the
nodule aspirate can confirm gout.
Tophi or uric acid deposits are found in :
- cartilage
- synovial membrane (membrane covering
the joints)
- tendons
- soft tissues

Tophi can occur in various organs,
including:
- finger
- hand
- knee
- foot
- ear
- elbow
- Achiles tendon
- spine
- internal organs, such as kidney and
even heart

http://www.skinsight.com/adult/gout.htm
The skin over the tophi lumps can form
ulcers and secrete pus.
In advanced chronic gout, damage to the
kidney caused by uric acid deposit can cause kidney failures. Other conditions,
such as hypertension (high blood pressure), albuminuria (abnormal presence of
albumin protein in the urine indicating kidney disease), and urolithiasis
(urinary stone in the urinary tract) can also develop.

http://malformalady.tumblr.com/post/20428297781/tophaceous-gout-is-a-chronic-form-of-gout-wherein

http://www.inpodiatrygroup.com/gout.html

http://www.healthinplainenglish.com/health/musculoskeletal/gout/index.htm
Surgical removal of the the
uric acid deposit.
|
American College of Rheumatology Preliminary
Criteria of Acute Arthritis of Primary Gout r Gout |
|
Gout may be diagnosed if one of the
following criteria is present: 1. Monosodium urate crystals in synovial fluid 2. Tophi
confirmed with crystal examination 3. At least six of the following findings: · Asymmetric swelling
within a joint on a radiograph · First
metatarsophalangeal joint is tender or swollen · Hyperuricemia · Maximal
inflammation developed within one day · Monoarthritis attack · More than one acute
arthritis attack · Redness observed
over joints · Subcortical cysts
without erosions on a radiograph · Suspected tophi · Synovial
fluid culture negative for organisms during an acute attack · Unilateral
first metatarsophalangeal joint attack · Unilateral tarsal
joint attack |
|
|
Laboratory and
instrumental findings
Hyperuricemia is a
serum uric acid (SUA) level consistently higher than 6.8 mg/dL. Hyperuricaemia
is arbitrarily denned as a serum uric acid level greater than two standard
deviations from the mean, i.e. above 7.0mg/dl (0.42mmol/l) in adult males and
6.0mg/dl (0.36 mmol/l) in adult females.
The serum urate
level is usually raised but it is important to appreciate that this does not
prove the diagnosis, because asymptomatic hyperuricaemia is very common. Also note that asymptomatic hyperuricemia does not need to be
managed when you have pseudogout.
Comparison of Gout and Pseudogout
|
|
Gout |
Pseudogout |
|
Ratio of men to women |
7:1 |
1:1.5 |
|
Age group affected |
Men >40 years old Postmenopausal women |
Elderly |
|
Serum urate |
Elevated |
Normal |
|
Joints involved |
First metatarsophalangeal (MTP) joint, insteps,
knees, wrists, fingers, olecranon bursae |
Knees, wrists, ankles |
|
Involvement of first MTP(podagra) |
Common |
Rare |
|
Tophi |
Present |
Rare tophi-like deposits |
|
Radiographic findings |
Erosions with overhanging edges |
Chondrocalcinosis |
|
Crystals |
Needle-shaped, strong negative birefringence |
Rhomboid-shaped, weakly positive birefringence |
The criterion
standard in the diagnosis of gout is the analysis of synovial fluid samples obtained with
aspiration. Wet mounts of the synovial fluid in gout reveal negatively
birefringent urate crystals. Also, the synovial fluid usually reveals an
inflammatory process, with a white blood cell count in the range of
7,000-10,000 x 103 per microliter. Synovial fluid findings can help in making
differential diagnose:
Differential Diagnosis of Acute Gout
|
Diagnosis |
Joint distribution |
Synovial fluid findings |
|||
|
WBC count* |
Gram stain/culture |
Synovial fluid crystals† |
Radiography findings |
||
|
Gout |
Lower extremities: metatarsophalangeal, midtarsal, or knee
joints; initial attacks may be less common in upper extremities |
2,000 to 50,000 per mm3(2 × 109to 50
× 109 per
L) |
Negative |
Needle shaped, negative birefringence |
Acute: asymmetric swelling |
|
Chronic: periarticular erosions with overhanging edges |
|||||
|
Pseudogout (calcium pyrophosphate deposition disease) |
Knee, wrist, or first metatarsophalangeal |
2,000 to 50,000 per mm3 |
Negative |
Rhomboid shaped, weak positive birefringence |
Soft tissue swelling, chondrocalcinosis (calcification of
cartilage) |
|
Septic arthritis |
Knee is most commonly involved (may be any joint distribution) |
< 50,000 per mm3 |
Positive |
No crystals |
Joint effusion; radiography results otherwise normal early in
the disease |
NOTE: This table applies to
immunocompetent patients.
WBC = white blood cell.
*—The synovial fluid
WBC count should not be used alone to exclude infection.
†—Septic
arthritis may coexist with crystalline arthritis.
Radiographic Appearance
Plain-film
radiography may be used to evaluate gout; however, findings generally do not
appear until after at least 1 year of uncontrolled disease. Bone scanning may
also be used to examine gout; the key finding on bone scans is an increased
radionuclide concentration at affected sites.
|
Early-phase 1 findings in gout are limited to the soft
tissues. The typical finding is an asymmetric swelling around the affected
joint. Another finding that may be evident in the early phase of gout is
edema of the soft tissues around the joints. In a patient who has had multiple
episodes of gouty arthritis in the same joint, a cloudy area of increased
opacity may be seen on plain-film radiographs |
|
|
In the intermediate phase 2 of
gout, the earliest bony changes appear. Most commonly, the bony changes
initially appear in the first metatarsophalangeal joint area. These early
changes are generally seen outside the joint or in the juxta-articular area.
These intermediate-phase findings are often described as punched-out lesions,
which can progress to become sclerotic as they increase in size. Fractures
may be present in affected areas in severe cases of intermediate-phase gout. |
|
|
|
In late - phase 3 gout, the hallmark findings are numerous
interosseous tophi. |




http://www.aafp.org/afp/2007/0915/p801.html
Another appearance showing multiple erosion locations including first MTP,
base of third and fourth metacarpals, and possibly the head of the fifth
metacarpal and second proximal phalanx.
High-resolution CT. The advantages are
superiority to plain radiography for detecting early disease and tophi changes.
The disadvantages are high cost, high radiation exposure, moderate
availability, and a lack of specificity.
DECT. The advantages of this
newer diagnostic study are sensitivity and specificity for urate deposits,
especially those in soft tissue and bone structures.9 The
disadvantages: expensive and high radiation exposure.
MRI. This modality detects
tophi with representative decreased signal in both T1- and T2-weighted images
with variable enhancement.8 MRI is a useful examination when
the presence of tophi is suspected but not proven. MRI often demonstrates
greater-sized tophi than expected or appreciated on physical examination. The
advantages are superiority to plain radiography in early detection and
characterization of tophi and no radiation exposure. The disadvantages are less
benefit than CT scanning, a lack of specificity for tophi, moderate
availability, and high expense.

http://www.sciencedirect.com/science/article/pii/S0033838908001528
Management
Lifestyle factors
Dietary factors are
thought to play a significant role in the increasing prevalence. Obesity is the
commonest comorbidity that highlights the importance of addressing diet .
Despite long-standing links between diet and gout, only recently have studies
described protective or causative components. Higher intakes of alcohol
(especially beer), fructose (found in many soft drinks), meat and seafood
increase risk, whereas coffee [23], dairy products and low BMI are protective.
Both vitamin C and cherries lower sUA levels.
Traditionally, patients were advised to adopt low purine diets, avoiding meat,
seafood and purine-rich vegetables. Such diets are broadly unappealing and
rarely followed. A calorie-restricted diet with low carbohydrate (40% of
energy), high protein (30% of
energy) and unsaturated fat (30% of
energy) should be recommended. Although life style
modification is unlikely to significantly reduce sUA, it carries additional
benefits in controlling other components
of metabolic syndrome associated with gout.
Hyperuricaemia should also trigger
assessment for common associated disease, principally those of metabolic
syndrome, present in 63% of men with gout. Hypertriglyceridaemia, hypertension,
type 2 diabetes, hyperlipidaemia and obesity are the features of metabolic
syndrome, which is strongly associated with cardiovascular disease risk. A key
physiological change in metabolic syndrome is insulin resistance which
decreases renal clearance of UA. Gout is associated with insulin-resistance
syndrome, hypertension and hyperlipidaemia. Hyperuricaemia itself may even be
an independent risk factor for cardiovascular disease.
Treatment of acute gout
Following lifestyle
advice, there are three main aspects to gout management; acute flare treatment,
depletion of excess UA stores and sUA reduction.The algorithms summarize the
current medical treatment of acute gout and chronic treatment. Products that
are currently licensed for the chronic management of gout in the UK are used as
the first- and second-line treatments and those that are used on a named
patient basis can be used thereafter. National institute of clinical excellence
(NICE) guidance has indicated that febuxostat can only be used in patients who
have contraindications to, or are intolerant of allopurinol.
Fig. 1. Algorithm for the
medical treatment of acute gout.

PPI: proton pump inhibitor.
The aim of treating
attacks is to promptly and safely resolve pain. Joint aspiration is not
essential to diagnose acute gout, but remains the gold standard and should be
performed if there is any uncertainty in diagnosis or suspicion of sepsis.
Without treatment, the pain of an acute attack will last for at least a week. Time
from treatment to termination is the only guide to judge the efficacy of acute
treatments as few placebo-controlled trials exist. In addition to
pharmacological agents, affected joints should be rested for 1–2 days and
treated with ice which has a significant analgesic effect.
NSAIDs (conventional and COX-2
inhibitors)
NSAIDs are the most
commonly used first-line treatment in an acute flare. Maximum doses of an NSAID
should be commenced quickly, tapering 24 h after complete symptom resolution.
Head to head NSAID studies show few differences amongst agents. NSAIDs have
many adverse effects (AEs) and should be avoided in gastrointestinal ulcer
disease, bleeding or perforation, renal insufficiency, heart failure and those
taking oral anti-coagulants. AEs are increased in the elderly and
co-administration of a proton pump inhibitor should be considered. When
contemplating NSAIDs, pre-morbid conditions and drug history should be taken
into account on an individual patient basis and any current national guidance
adhered to.
Colchicine
Colchicine is an
alkaloid derived from the autumn crocus (Colchicum autumnale), and first
used in the 6th century AD by Alexander of Tralles. The earliest mechanism
described is the ability of colchicine to block microtubule assembly in
neutrophils reducing phagocytosis and transport of MSU crystals. Colchicine
also affects neutrophil migration into joints by reducing adhesion molecules on
endothelial cells and neutrophils in response to IL-1 or TNF-α. More
recently, it has been demonstrated that colchicine also reduces NALP3
inflammasome-driven caspase-1 activation by microtubule inhibition which
decreases MSU delivery.
Corticosteroids
Corticosteroids act
on the cytosolic glucocorticoid receptor to alter gene expression. Steroids also
have non-genomic effects mediated by the cytosolic glucocorticoid receptor,
membrane-bound glucocorticoid receptor and additional interactions with
cellular membrane proteins . Corticosteroids are a good alternative where NSAID
and colchicine cannot be used or in refractory cases.
IL-1 inhibitors
Anakinra, an IL-1
receptor antagonist, is a new treatment in development. The therapeutic basis
for this treatment stems from the discovery that MSU crystals stimulate the
inflammasome leading to IL-1β secretion. In current experiments, IL-1
inhibitors prevent IL-1 secretion via this mechanism and also block IL-1
secretion by marcophages via a TLR-dependent mechanism . IL-1 inhibition has
also shown success in treating hereditary autoinflammatory syndromes, where
mutations in theNALP3 gene result in spontaneous activation of the
NALP3 inflammasome.
Chronic management
Fig. 2. Algorithm for the medical treatment of chronic gout.

Currently, no evidence suggests that asymptomatic hyperuricaemia should be treated, although lifestyle advice should be offered. Urate lowering therapy (ULT) is indicated to treat recurrent attacks, arthropathy, tophi, UA renal lithiasis and radiographic evidence of gout. There is currently no defined point at which to initiate ULT. ULT can be divided into uricostatic agents that decrease UA production, uricosuric agents that increase renal excretion or uricolytic agents that metabolise UA. Figure 3 summarizes how ULTs exert their effects.
Fig. 3. Summary of the final
part of purine metabolism and site of drug action (XO = xanthine oxidase).

The therapeutic goal
is to prevent MSU crystal formation by following BSR or EULAR MSU targets of ⩽ 0.30 mmol/L or 0.36 ⩽ mmol/L. These values within the normal
range of 0.20–0.42 mmol/l quoted by most British laboratories and can cause
confusion. Sustained control of sUA below target levels gives good long-term
clinical outcomes and decreases flare frequency. However, the optimum target of
sUA is unknown and could vary in different patient groups.
Prophylaxis
Prophylaxis against
acute attacks should be given when ULT is initiated, either with an NSAID or
colchicine. If no prophylaxis is initiated, 77% of the patients experience
flares in the first 6 months of commencing allopurinol. One must minimize
flares on initiation of ULT, as this is a commonly cited reason for
non-concordance.
Colchicine provides effective prophylaxis
at low dose, and fewer subjects experience diarrhoea as a side effect than at
treatment dose. Results from RCT indicate prophylactic colchicine 600 µg
twice a day should be used for at least 3 months and up to 6 months upon
initiating ULT, as this significantly reduces flare frequency and severity.
Uricostatic agents, xanthine
oxidase inhibitors
Allopurinol
For the past 30 years, allopurinol has
been the mainstay of chronic treatment and accounts for 90% of ULT . It is an
effective agent and there is a significant inverse relationship between
allopurinol dose and sUA . Allopurinol reduces sUA by inhibiting xanthine
oxidase (XO) thereby preventing xanthine, a product of purine catabolism, being
converted into UA as shown in Fig. 3. It should be commenced at 100 mg daily
and increased by 100 mg every 1–2 weeks titrated against sUA and
creatinine clearance (maximum dose is 900 mg) . AEs of allopurinol include rash
(2%), vasculitis, eosinophilia, life-threatening hypersensitivity reaction,
hepatitis, decreased renal function and bone-marrow suppression. Allopurinol
requires reduced dosing in renal impairment, this being its route of excretion.
Oxypurinol, the active metabolite of allopurinol, is an alternative in
allopurinol allergy, but there is a 40% chance of cross reactivity.
Febuxostat
Febuxostat is a new agent which
selectively inhibits XO independent of the redox state and does not affect
other enzymatic pathways in purine/pyrimidine metabolism. Febuxostat is
extensively metabolized by conjugation via uridine diphosphate
glucuronosyltransferase and to lesser extent by the cytochrome P450 system. No
dose reduction in moderate renal impairment (or moderate hepatic impairment) is
required. Other advantageous properties include no interaction with
warfarin and a safe alternative in patients with allopurinol
allergy. Febuxostat appears to be well tolerated, the most common AEs being
abnormal liver function tests (LFTs). Others include diarrhoea,
joint-related/musculoskeletal/connective tissue symptoms, flushing, dizziness,
confusion, myalgia and tachycardia.
Uricosuric agents
These drugs enhance renal clearance of
urate and were first introduced at the end of the 19th century. They are used
in <15% of gout patients. Benzbromarone, sulphinpyrazone
and probenecid all directly inhibit URAT-1 and therefore reduce urate
reabsorption. Uricosurics are contraindicated in urate nephropathy or history
of acute nephrolithiasis. An increased fluid input and output is therefore
recommended for all patients. UA stone formation is not common; however, the
most important risk factor for UA crystallization and stone formation is a low
urine pH (<5.5), rather than an increased urinary UA excretion. To prevent a
low urinary pH and decrease the risk of nephrolithiasis, one can alkalinize the
urine using potassium citrate or bicarbonate, with the goal of increasing urine
pH to values >6.0, and up to 7.0. Usually, advice from a renal physician
should be sought and all patients should be encouraged and instructed about
maintaining urine volumes of at least 2 l per day.
Benzbromarone
Benzbromarone is metabolized by cytochrome
P450 and was withdrawn from widespread use because of serious hepatotoxicity.
It has been estimated that the risk of hepatotoxicity is 1 : 17 000 taking into
account four published cases and 11 cases reported by Sanofi-Synthelabo (Paris, France). nBenzbromarone is a highly effective drug with 100% of
the patients achieving target urate levels of <6 mg/dl in a trial showing
comparable efficacy to allopurinol . Benzbromarone doses of 50–200 mg
daily are used and generally well tolerated, although regular LFT monitoring is
essential. Benzbromarone additionally inhibits SLC2A9, and is the only
uricosuric that is effective in moderate renal impairment. It is particularly
useful where allopurinol is contraindicated or not tolerated, such as in the
management of renal transplant patients. This suggests that benzbromarone is a
better choice following treatment failure or AEs with allopurinol.
Sulphinpyrazone
Sulphinpyrazone inhibits prostaglandin
synthesis much like the NSAIDs and therefore its AEs are similar including
gastro-intestinal ulceration, acute renal failure, fluid retention and rarely
elevation of liver enzymes and blood disorders. Sulphinpyrazone 200–800
mg daily in divided doses is used. It has no efficacy in renal impairment and
adverse reactions make its clinical use difficult.
Probenecid
Probenecid can be effective as an add-in
therapy when allopurinol alone is insufficient, but is ineffective in renal
impairment. Divided doses of 0.50–2.0 g are used but it is rarely
utilized due to difficulties with supply.
Uricolytics
Humans unlike nearly all mammals have
mutations in the genes encoding the enzyme uricase. The human uricase gene
underwent two separate mutations that independently resulted in truncation of
gene transcription. This decreased uricase function, but may have increased
antioxidant activity, increased intelligence and improved the ability of humans
to retain salt. The action of uricase converts urate to allantoin, which is 10
times more soluble and thus more readily excreted. During the past seven
decades, there have been numerous attempts to administer uricase; however, it
now appears that this exogenous enzyme has been successfully converted into an
effective drug.
Rasburicase
In 1996, rasburicase was developed by
recombinant DNA technique from a genetically modified strain of Saccharomyces
cerevisiae. The efficacy of rasburicase in prevention and treatment of tumour
lysis syndrome (TLS) has been well demonstrated despite its cost. However,
allergenicity and development of antibodies compromise its effectiveness, the
risk of which increases with repeated use . Rasburicase is given IV at a dose
of 0.20 mg/kg for 5–7 days to treat TLS.
Others
Losartan, an
angiotensin II receptor antagonist used for hypertension, and fenofibrate, a
fibric acid derivative used in hyperlipidaemia, both have uricosuric actions
and reduce sUA . The action on sUA is not a class effect for either drug and
neither is licensed in treating gout. Losartan inhibits URAT-1, whereas fenofibrate can down-regulate
the expression of the inducible COX-2 enzyme, but its exact mechanism is less
well understood.
This effect of losartan and fenofibrate on
sUA is particularly beneficial, given the frequent co-existence of hypertension
and hyperlipidaemia with gout. Preferential use of these agents when treating
comorbidities of gout is recommended. Together, these agents can decrease sUA
by 40%; however, fractional UA clearance is increased by 110% and there is a
risk of urolithiasis.

Systemic lupus erythematosus (SLE)
Systemic
lupus erythematosus (SLE) is a chronic inflammatory
disease of unknown cause which can affect the skin, joints, kidneys, lungs,
nervous system, serous membranes and/or other organs of the body. Distinct
immunologic abnormalities, especially the production of a number of antinuclear
antibodies, are another prominent feature of the disease. The clinical course
of SLE is characterized by periods of remissions and chronic or acute relapses.
Women, especially in their 20s and 30s, are affected more frequently than men.
Treatment is based on preventive measures, reversal of inflammation, prevention
of organ impairment, and alleviation of symptoms.
History
The term
‘lupus’ was first used during the Middle Ages to describe erosive skin lesions
evocative of a ‘wolf’s bite’. In 1846 the Viennese physician
Ferdinand von Hebra (1816–1880) introduced the butterfly metaphor to
describe the malar rash. He also usedthe term ‘lupus erythematosus’ and published the first illustrations in his Atlas
of Skin Diseases in 1856. Lupus was first recognised as asystemic disease with
visceral manifestations by Moriz Kaposi (1837–1902). Th e systemic form
was further established by Osler in Baltimore and Jadassohn in Vienna. Other important
milestones include the description of the false positive test forsyphilis in
SLE by Reinhart and Hauck from Germany (1909);the description of the endocarditis lesions in SLE
by Libman and Sacks in New York (1923);
Epidemiology
International statistics
The highest rates of
prevalence have been reported in Italy, Spain, Martinique, and the United
Kingdom Afro-Caribbean population. Although the prevalence of SLE
is high in black persons in the United Kingdom, the disease is rarely reported in blacks in Africa,
suggesting that there may be an environmental trigger, as well as a genetic
basis, for disease in the UK population. The annual incidence of SLE
averages 5 cases per 100,000 population. The Centers for Disease Control and
Prevention (CDC) estimates a range between 1.8 and 7.6 per 100,000 persons per
year in the continental United States.
The Lupus Foundation of American estimates
prevalence to be up to 1.5 million cases, which
likely reflects inclusion of milder forms of this disease. The frequency of SLE
varies by race and ethnicity, with higher rates reported in blacks and
Hispanics. The incidence of SLE in black women is approximately 4 times higher
than that in white women. SLE is also more frequent in Asian women than in
white women.
Race-, sex-, and age-related
demographics
Worldwide, the
prevalence of SLE appears to vary by race. However, there are different
prevalence rates for people of the same race in different areas of the world.
The contrast between low reported rates of SLE in black women in Africa and
high rates in black women in the United Kingdom suggests that there are environmental influences In
general, black women have a higher rate of SLE than women of any other race, followed
by Asian women and then white women.

Female-to-male ratio
More than 90% of
cases of SLE occur in women, frequently starting at childbearing age. The use of exogenous
hormones has been associated with lupus onset and flares, suggesting a role for
hormonal factors in the pathogenesis of the disease. The risk of SLE development
in men is similar to that in prepubertal or postmenopausal women.
Interestingly, in men, SLE is more common in those with Klinefelter syndrome (ie, genotype XXY), further
supporting a hormonal hypothesis. In fact, a study by Dillon et al found that
men with Klinefelter syndrome had a more severe course of SLE than women but a
less severe course than other men.
The female-to-male
ratio peaks at 11:1 during the childbearing years. A correlation between age
and incidence of SLE mirrors peak years of female sex hormone production. Onset
of SLE is usually after puberty, typically in the 20s and 30s, with 20% of all
cases diagnosed during the first 2 decades of life. The prevalence of SLE is highest in
women aged 14 to 64 years. SLE does not have an age predilection in males,
although it should be noted that in older adults, the female-to-male ratio
falls. This
effect is likely due to loss of the estrogen effect in older females.
Etiology
Although the
specific cause of SLE is unknown, multiple genetic predispositions and
gene-environment interactions have been identified (see the chart in the image
below). This complex situation perhaps explains the variable clinical
manifestations in persons with SLE.
HLA = human leukocyte antigen; UV = ultraviolet light.
In systemic lupus
erythematosus (SLE), many genetic-susceptibility factors, environmental
triggers, antigen-antibody (Ab) responses, B-cell and T-cell interactions, and
immune clearance processes interact to generate and perpetuate autoimmunity.
SLE has a modest
recurrence rate in families: 8% of affected patients have at least one
first-degree family member (parents, siblings, and children) with SLE; this is
in contrast to 0.08% of the general population. In addition, SLE occurs in
both twins in 24% of identical twins and 2% of nonidentical twins, which may be
due to a combination of genetic and environmental factors. Some studies have
synthesized what is known about the mechanisms of SLE disease and genetic
associations. At least
35 genes are known to increase the risk of SLE. A genetic
predisposition is supported by 40% concordance in monozygotic twins; if a
mother has SLE, her daughter's risk of developing the disease has been
estimated to be 1:40, and her son's risk, 1:250.
HLA-A1, HLA-B8, and HLA-DR3 are more
common in persons with SLE than in the general population. The presence of the
null complement alleles and congenital deficiencies of complement (especially
C4, C2, and other early components) are also associated with an increased risk
of SLE.
Patients with SLE
have higher titers of antibodies to Epstein-Barr virus (EBV), have increased
circulating EBV viral loads, and make antibodies to retroviruses, including
antibodies to protein regions homologous to nuclear antigens. In patients with
SLE and EBV infection, the B cells are not primarily defective; rather, the
SLE/EBV phenomenon is due to a T-cell abnormality, which causes failure in
normal immunoregulation of the B-cell response. Viruses may stimulate specific
cells in the immune network. Chronic infections may induce anti-DNA antibodies
or even lupuslike symptoms, and acute lupus flares often follow bacterial
infections.
POTENTIAL ETIOLOGIC FACTORS
• Viruses (EBV)
• Hormones (estrogen)
• Genetic
predisposition (HLA B8)
• Drugs (e.g.,
procainamide)
↓
Loss of tolerance
↓
Polyclonal В cell hyper-reactivity
↓
AUTOANTIBODY PRODUCTION
(anti-double-stranded DNA, etc.)
↓
Immune complex formation in circulation and
tissues
↓
TISSUE INJURY
• Glomerulonephritis
• Vasculitis
• Serositis
• Arthritis
Environmental and exposure-related causes
of SLE are less clear. They potentially include the following:
· Silica dust and cigarette smoking may
increase the risk of developing SLE
· Administration of estrogen to
postmenopausal women appears to increase the risk of developing SLE.
· Breastfeeding is associated with a
decreased risk of developing SLE
· Photosensitivity is clearly a precipitant
of skin disease
· Ultraviolet light stimulates
keratinocytes, which leads not only to overexpression of nuclear ribonucleoproteins
(snRNPs) on their cell surfaces but also to the secretion of cytokines that
simulate increased autoantibody production.
Patogenesis
The pathogenesis of
lupus remains unclear although the concept of apoptosis goes some way to
explaining how the immune system may recognise predominantly intracellular
antigens. Autoantigens are released by necrotic as well as apoptotic cells.
Defects in the clearance of apoptotic cells have been described in SLE which
may lead to aberrant uptake by macrophages which then present the
previously intracellular antigens to T and B cells thus driving the autoimmune
process. Recent work has expanded these concepts and dissected out possible
defects in clearance of apoptotic bodies including complement deficiencies, defects
in macrophage handling and presentation
of these antigens to the immune system. The most striking recent studies have
demonstrated the development of autoantibodies years before the onset of clinical features
of SLE and the antiphospholipid syndrome (APS). Antinuclear antibodies occurre
earlier than antiDNA antibodies and a significant number of these patients had
a rise in the anti-DNA titres just prior to diagnosis. Interestingly, anti-Sm
and anti-RNP antibodies appeared shortly before diagnosis suggesting a
crescendo of autoimmunity resulting in clinical illness. This data also
suggests that autoantibodies alone do not necessarily result in clinical
disease and that other factors possibly genetic and environmental may be
important. It may be possible in the future to predict the onset of clinical
features of lupus by clinical assessment and monitoring the development of
various lupus autoantibodies.
Classification
The
nature of the disease
ü acute
ü subacute
ü chronic:
Ø recurrent arthritis,
Ø discoid lupus,
Ø Raynaud's syndrome,
Ø thrombocytopenic purpura syndrome,
Ø Sjogren syndrome
Stage
of activity
ü Active
Ø high
Ø moderate
Ø minimal
ü Moderate
ü Severe
CLINICAL MANIFESTATIONS
Musculoskeletal involvement
Joints
Arthralgia occurs in
about 90% of all patients with SLE. Characteristically, it is polyarticular,
symmetrical, episodic and flitting in nature. The patients’ symptoms
often exceed the objective clinical findings and usually there is no clinically
overt arthritis. Synovial effusions are uncommon and of small volume when they
do occur. However, approximately 10% of SLE patients do have a deforming
Jaccoud’s arthritis. In contrast to patients with rheumatoid arthritis,
the deformities are not usually associated with synovial hypertrophy or bony
erosions. In fact, tenosynovitis is more common than
erosive synovitis and is the cause of the
“swan-neck” deformities and ulnar deviation seen in the
Jaccoud’s arthritis of lupus. Examination of the synovial fluid usually
reveals a white cell count of less than 3000/mm3, predominantly mononuclear
cells. The fluid is often positive for rheumatoid factor and anti-nuclear
antibody
Muscles
Clinically obvious
muscle involvement has been reported in 30-50% of SLE patients. However,
myalgia, muscle weakness and tenderness, may be due to a variety of other
complications. Thus both corticosteroid and rarely chloroquine therapy may
cause a myopathy. In addition, myalgia may be induced by an adjacent
arthralgia, although only 5% of lupus patients have met the ACR criteria for
both SLE and polymyositis.
Dermatological involvement

Cutaneous lesions
may occur in up to 85% of SLE patients. The butterfly rash is
erythematous, often blotchy, and found
mainly over the malar bones and across the bridge of the nose.

Vasculitic skin lesions are usually found at the
nailfolds and finger tips or
on the extensor surface of the forearm.
When they occur around the malleoli, they may lead to tender, deep, leg ulcers
which can take months to heal.
Many SLE rashes are
exacerbated by ultraviolet light and indeed generalized lupus flares may follow exposure to direct sunlight
with inadequate protection. A
particularly photosensitive rash is subacute cutaneous lupus
erythematosus (SCLE) which is
often associated with anti-Ro antibodies.
Babies born to mothers with anti-Ro and/or
anti-La antibodies are at risk of neonatal lupus syndrome.
The deposition of
immunoglobulins at the dermal-epidermal junction in skin biopsies from patients
with lupus was first reported over 40 years ago. These immunoglobulins are usually
of the IgG or IgM isotype.
Approximately, 90% of biopsies from lupus skin lesions have such immunoglobulin deposits which usually
appear as a band along the dermal-epidermal junction, giving rise to the
name the “lupus band test”. In patients with SLE, deposition of
immunoglobulin and complement
may be found in clinically normal skin and is thus a useful adjunct to
diagnosis since no such
deposition is found in patients with discoid lupus or control subjects.
Lupus nephritis
More than 70% of
patients with SLE have renal involvement at some stage of their disease.
These descriptions allow
better communication between pathologists translating static images from
histology slides into meaningful descriptions of the huge variety of biopsy
appearances for clinicians. Of the different pathological classes, diffuse
proliferative glomerulonephritis (Class IV) has the worst
prognosis, resulting in 11-48% of patients
with end stage renal disease at 5 years.
International Society of
Nephrology/Renal Pathology Society 2003 classification of lupus nephritis
Class I
Minimal mesangial lupus
nephritis
Normal glomeruli by light
microscopy, but mesangial immune deposits by immunofluorescence
Class II
Mesangial proliferative lupus
nephritis.
Purely mesangial
hyper-cellularity of any degree or mesangial matrix expansion by light
microscopy, with mesangial immune deposits. May be a few isolated
sub-epithelial or sub-endothelial deposits visible by immunofluorescence or
electron
microscopy, but not by light
microscopy
Class III
Focal lupus nephritisa
Active or inactive focal,
segmental or global endo- or extra-capillary glomerulonephritis involving
<50% of all glomeruli, typically with focal sub-endothelial immune deposits,
with or without mesangial alterations
Class III (A)
Active lesions: focal
proliferative lupus nephritis
Class III (A/C)
Active and chronic lesions:
focal proliferative and sclerosing lupus nephritis
Class III (C)
Chronic inactive lesions with
glomerular scars: focal sclerosing lupus nephritis
Class IV
Diffuse lupus nephritis
Active or inactive diffuse,
segmental or global endo- or extra-capillary glomerulonephritis involving 50%
of all glomeruli, typically with diffuse sub-endothelial immune deposits, with
or without mesangial alterations. This class is divided into diffuse
segmental(IV-S) lupus nephritis when 50% of the involved glomeruli have
segmental lesions, and diffuse
global (IV-G) lupus nephritis when 50% of the involved glomeruli have global
lesions. Segmental is defined as a glomerular lesion that involves less than
half of the glomerular tuft. This class
includes cases with diffuse
wire loop deposits but with little or no glomerular proliferation
Class IV-S (A)
Active lesions: diffuse
segmental proliferative lupus nephritis
Class IV-G (A)
Active lesions: diffuse global
proliferative lupus nephritis
Class IV-S (A/C)
Active and chronic lesions:
diffuse segmental proliferative and sclerosing lupus nephritis
Active and chronic lesions:
diffuse global proliferative and sclerosing lupus nephritis
Class IV-S (C)
Chronic inactive lesions with
scars: diffuse segmental sclerosing lupus nephritis
Class IV-G (C)
Chronic inactive lesions with
scars: diffuse global sclerosing lupus nephritis
Class V
Membranous lupus nephritis
Global or segmental
sub-epithelial immune deposits or their morphologic sequelae by light
microscopy and by
immunofluorescence or electron
microscopy, with or without mesangial alterations
Class V lupus nephritis may
occur in combination with class III or IV in which case both will be diagnosed
Class V lupus nephritis show
advanced sclerosis
Class VI
Advanced sclerosis lupus
nephritis
90% of glomeruli globally
sclerosed without residual activity
A Indicate the proportion of
glomeruli with active and with sclerotic lesions.
B Indicate the proportion of
glomeruli with fibrinoid necrosis and/or cellular crescents.
Indicate and grade (mild,
moderate, severe) tubular atrophy, interstitial inflammation and fibrosis,
severity of
arteriosclerosis or other
vascular lesions.
Lungs
The
immunosuppressive therapy required by many SLE patients predisposes them to
concurrent infection. The lungs are a frequent target for this
“secondary” infection and bacteria (including tubercule bacilli),
viruses and fungi may all cause pneumonia in lupus patients.Parenchymal
alterations, attributable to SLE itself, have been described in 18% of
patients. These patients had interstitial fibrosis, pulmonary vasculitis and
interstitial pneumonitis. However, many non-specific pulmonary lesions
previously attributed to SLE, such as alveolar haemorrhage alveolar wall
necrosis, oedema and hyaline membranes, are probably secondary to factors such
as intercurrent infection, congestive heart failure, renal failure and oxygen
toxicity.In the relatively few cases studied, immune complex deposition has
been closely correlated with histological evidence of inflammatory lesions in
the pleural (and pericardial) membrane.
Abnormal pulmonary function tests, notably
diminished total lung capacity and flow rates, in clinically mild patients with
dyspnoea, poor diaphragmatic movement, basal crepitations and occasionally
cyanosis and clubbing, are found in up to 50% of SLE patients. A similar
proportion of SLE patients may have an acute lupus pneumonitis with a
mononuclear cell infiltrate detectable in the alveolar septae. These patients
frequently complain of dyspnoea, pleuritic chest pain and coughs. Haemoptysis
is less common and true pulmonary haemorrhage from necrotizing alveolar
capillaritis is rare. Pleural effusions may be found in about half of these
patients (and in other SLE patients especially during generalized disease
flares). The effusions are normally small to moderate in size and are usually
exudates (i.e. protein content >3 g/100 ml). They are rarely haemorrhagic
and usually have a glucose concentration double that found in rheumatoid
effusions (normally, 20 mg/100 ml or less).
Heart
Pericardium
Abnormalities of the
electrocardiogram, notably of the T wave, are the most frequent
manifestation. A pericardial rub may be
more common than a significant pericardial effusion. Histological abnormalities
vary from occasional foci of fibrinoid degeneration and inflammatory cell
infiltrates to far more extensive lesions. Adhesive chronic pericarditis and
very large effusions causing tamponade are very rare.
Myocardium
Whilst true
myocardial involvement is less frequent than pericardial disease, prolongation
of the PR interval (approximately 10%), fibrinoid degeneration, myocardial
infarction and coronary stenosis due to arteritis are occasionally seen. New
imaging techniques such as cardiac MRI suggest that myocardial involvement may
be more common than previously thought.There is increasing evidence that
premature accelerated atherosclerosis considerably increases the risk of
cardiovascular events in patients with SLE and this is described in a separate
module of this course.
Valves
Systolic murmurs are
frequently heard in around 30% of SLE patients. However, they probably reflect
the hyperdynamic circulation consequent upon the anaemia often found in these
individuals. In contrast, diastolic murmurs are uncommon. Libman-Sacks
endocarditis has long been described as a feature of SLE. Although found in up
to 50% of autopsied cases, it rarely
causes clinically significant lesions. Histologically, the lesions are small
(1-4 mm) vegetations (verrucae) comprising
proliferating and degenerating valve tissue with fibrin and platelet thrombi.
They are most frequently found adjacent to the edges of the mitral and
tricuspid valves. aPL may contribute to the development of Libman-Sacks
endocarditis and studies suggest that there is a selective deposition of aPL
and complement within the walls of the small junctional vessels in the active
portions of the verrucous endocardial lesions.
Central nervous system lupus
The ACR
classification criteria for central nervous system (CNS) lupus has changed
considerably from seizures and psychosis.
The ACR nomenclature now includes 19 different syndromes that are classifiable
. An emerging concept is the distinction between CNS manifestations due to
lupus and those due to the APS. A wide variety of neuropsychiatric
manifestations attributable to APS have been described including strokes,
seizures, movement disorders, transverse myelopathy, demyelination syndromes,
transient ischaemic attacks, cognitive dysfunction, visual loss and headaches
including migraine.
Neuropsychiatric syndromes
observed in SLE.
Central nervous system:
ü Aseptic meningitis
ü Cerebrovascular disease
ü Demyelinating syndrome
ü Headache (including migraine and benign
intracranial hypertension)
ü Movement disorder (chorea)
ü Myelopathy
ü Seizure disorders
ü Acute confusional state
ü Anxiety disorder
ü Cognitive dysfunction
ü Mood disorder
ü Psychosis
Peripheral nervous system:
ü Acute inflammatory demyelinating
polyradiculoneuropathy (Guillain-Barré syndrome)
ü Autonomic disorder
ü Mononeuropathy, single/multiplex
ü Myasthenia gravis
ü Neuropathy, cranial
ü Plexopathy
ü Polyneuropath
Laboratory diagnosis of CNS lupus can be
difficult. Abnormal electroencephalograms occur in about 70% of patients with
neurologic complaints and usually show diffuse slowing or focal abnormalities.
Cerebrospinal fluid (CSF) shows elevated protein levels in 50% and increased
mononuclear cells in 30% of patients; oligoclonal bands and increased Ig
synthesis may be found. Lumbar puncture is recommended when the diagnosis of
CNS lupus is in doubt or when infection is a possible cause of symptoms.
Magnetic resonance imaging (MRI) with contrast is the most sensitive
radiographic technique to detect acute and chronic lesions of SLE; changes are
often nonspecific. Patients with focal neurologic lesions are more likely to
have positive MRI scans than those with diffuse manifestations. Computed
tomography (CT) scans are useful to rule out bleeding or mass lesions, if
indicated. Angiograms can detect vasculitis and vascular occlusions or emboli;
they cannot visualize vessels smaller than 50 um; lupus vasculitis usually
involves smaller vessels. Laboratory measures of disease activity often do not
correlate with neurologic manifestations. Neurologic problems (with the exception
of deficits resulting from large infarcts) usually improve with
immunosuppressive therapy and/or time; recurrences are seen in approximately
one-third of patients.
Gastrointestinal System
Common
gastrointestinal (GI) symptoms include nausea, diarrhea, and vague discomfort.
Symptoms may result from lupus peritonitis and may herald a flare of SLE.
Vasculitis of the intestine is the most dangerous manifestation, presenting
with acute crampy abdominal pain, vomiting, and diarrhea. Intestinal perforation
can occur and usually requires immediate surgery. Patients with
pseudoobstruction have abdominal pain; x-rays show dilated loops of small bowel
which may be edematous; surgery should be avoided unless frank obstruction is
present. Glucocorticoid therapy is useful for all these GI syndromes. Some
patients have GI motility disorders similar to those in scleroderma; they are
not benefited by steroids. Acute pancreatitis occurs and can be severe,
resulting from active SLE or from therapy with glucocorticoids or azathioprine.
Elevated amylase levels may reflect pancreatitis, salivary gland inflammation,
or macroamylasemia. Elevated serum transaminase levels are common in patients
with active SLE but are not associated with significant hepatic damage; they return
to normal as the disease is treated.
Ocular Manifestation
Retinal vasculitis
is a serious manifestation; blindness can develop over a few days, and
aggressive immunosuppression should be instituted. Examination shows areas of
sheathed, narrow retinal arterioles and cytoid bodies (white exudates) adjacent
to vessels. Other ocular abnormalities include conjunctivitis, episcleritis,
optic neuritis, and the sicca syndrome.
Esophagus
Lupus patients
occasionally complain of dysphagia or odynophagia. This can be multifactorial
from hypomotility, from reflux disease, or from candidiasis
from immunosuppression. If the symptoms are severe, they deserve a regular
dysphagia evaluation with motility studies, x-rays, and maybe an endoscopy.
Although treatment is directed at the cause, motility drugs are no longer
favored due to their arrythmogenic potential. Antireflux medications or
antifungals are used when appropriate.
Abdomen
Abdominal pain is a
diagnostic challenge in SLE and is probably one of the most clinically
threatening GI manifestation to be aware of. Min and colleagues looked at
causes of acute abdominal pain in SLE patients in emergency departments (EDs).
They documented that 59.1% of visits to the ED by SLE patients were from pain
due to ischemic bowel disease. The other causes were splenic infarcts, renal
venous thrombosis, pancreatitis, serositis, upper GI bleeds, pelvic
inflammatory disease, and ectopic pregnancy. Peptic ulcer disease with
perforation also manifested as an acute abdomen in a small number of patients
with SLE and concomitant NSAID use. Treatment of acute abdominal
pain is directed at the cause, with appropriate medical or surgical management
of the presenting manifestation.
Intestines
In the bowel, SLE
can manifest with vasculitis, malabsorption, or dysmotility. Mesenteric vasculitis in
lupus can manifest as an acute abdomen with fever, nausea, vomiting, diarrhea,
and rectal bleeding or with the characteristic mesenteric ischemic pain related
to meals. The mesenteric involvement can be attributed to either a lupus flare
or antiphospholipid antibodies. Suspicion based on a clinical, angiographic, or
CT examination of mesenteric vasculitis without bowel perforation warrants an
evaluation by a rheumatologist and a possible aggressive therapeutic approach
with intravenous steroids with or without other cytotoxic agents, besides the
routine treatments with nothing by mouth, IV fluids, cultures, and
broad-spectrum antibiotics. If there is intestinal perforation
from vasculitis, surgery is the first option followed by cautious start of
steroids and cytotoxic agents in the postoperative period. Malabsorption in the
form of a protein-losing enteropathy in lupus is uncommon and manifests with
diarrhea, abdominal pain, and anasarca. The enteropathy might respond to
steroids with or without cytotoxic drugs.
Pancreas
Pancreatitis in
lupus is uncommon and could occur in a setting of high SLEDAI scores,
antiphospholipid antibody syndrome, and probable steroid use. The more likely causes, as
in any other setting, are gallstones, alcohol, and hypertriglyceridemia.
Treatment is the same as for pancreatitis from any other cause and includes
nothing by mouth, IV fluids, withholding causal drugs, and, rarely, use of
steroids if the cause is established by exclusion.
Liver
Drugs, viruses,
fatty infiltration, or congestion have been implicated as more common causes of
liver enzyme abnormalities in SLE patients. Hepatitis from
lupus (lupus hepatitis), although uncommon, manifests as a mild elevation in
liver enzymes (aspartate transaminase [AST], alanine transaminase [ALT)],
lactate dehydrogenase [LDH], alkaline phosphatase), usually in a setting of
active lupus. Such biochemical liver abnormalities from an SLE flare have a
tendency to reverse with steroids. Lupoid hepatitis is a separate entity and is
considered a subset of chronic active autoimmune hepatitis, where the liver is
the main organ of involvement. Patients with lupus hepatitis and lupoid
hepatitis can have arthralgias, hypergammaglobulinemia, and positive ANAs. Serologic differentiation
may be possible at times and in general involves the presence of
anti–ribosomal P and dsDNA autoantibodies in lupus hepatitis versus
anti–smooth muscle and auto–liver-kidney-mitochondrial (LKM)
antibodies in lupoid hepatitis. Definite differentiation is only possible on
histology, which shows a lobular involvement in lupus hepatitis versus
rosetting of liver cells and dense lymphoid infiltrate in lupoid hepatitis.
Haematological abnormalities
Red blood cells
A normochromic,
normocytic anaemia is frequently found in SLE patients, with concomitant low
levels of both the serum iron and iron binding capacity. This abnormality
appears to be related, as in other diseases, to chronic inflammation and
shunting of elemental iron from erythroblasts to macrophages.Iron-deficiency
anaemia may be induced by non-steroidal anti-inflammatory drugs, which can
cause gastrointestinal haemorrhage. Excessive blood loss from menorrhagia,
sometimes related to severe thrombocytopenia, may have the same effect.
Haemolytic anaemia as detected by the Coombs’ test is another rare
feature of SLE. Autoimmune thrombocytopenia occasionally manifests
simultaneously with haemolytic anaemia: this
condition is known as Evan’s
syndrome.
Platelets
Two forms of thrombocytopenia
(platelet count < 100 x 109/l) are found in SLE. Firstly, it may be
encountered in a chronic form, generally associated with mild disease.
Secondly, it may occur in an acute form, similar to idiopathic autoimmune
thrombocytopenic purpura. This latter association is with disease carrying a
greater morbidity and mortality. Platelet destruction appears to be mediated by
anti-platelet antibodies and aPL are also associated with thrombocytopenia as
well as with thrombosis.
White blood cells
Persistent
leucopenia (< 4.0 x 109/l) is one of the ACR criteria for the classification
of SLE. It probably results from a combination of destruction of white cells by
autoantibodies, decreased marrow production, increased or marginal splenic
pooling, and complement activation. It should also be noted that the
immunosuppressive drugs used in the treatment of SLE may cause a marked
leucopenia.
Serological abnormalities
The serum from SLE
patients may bind to an extensive array of molecules including nucleic acids
(antinuclear antibodies) and phospholipid binding proteins (lupus
anticoagulant, anticardiolipin antibodies, β2 glycoprotein 1 antibodies).
Antibodies may also be detected against diverse cells including leukocytes,
erythrocytes, platelets and neurones. In addition to these autoantibodies,
numerous other abnormalities are evident, including the LE cell phenomenon,
hypocomplementaemia, elevated levels of acute phase proteins, gamma globulins
and circulating immune complexes.
Non-specific features
Fever,
lymphadenopathy, hair loss and Raynaud’s phenomenon are all commonly
found in SLE patients. Fever in lupus patients may be striking and often
requires extensive investigation to exclude concurrent infection, although a
normal CRP in this context usually suggests a low likelihood of sepsis.
Lymphadenopathy may also be dramatic in SLE, to such an
extent that lymph node biopsy may have to be performed to exclude malignancy.
Some patients seem more prone to this feature than others and in this group the
degree of lymphadenopathy may reflect general disease activity.
Splenomegaly occurs in about 10% of patients.
The clinical diagnosis of SLE hinges on
careful and very thorough assessment of the
presenting clinical features, examination
of all the organ systems and selected investigations.
Clinical symptoms
Symptoms often occur
intermittently and cumulatively over many months and years. Oral ulcers,
arthralgia, hair fall, Raynaud’s phenomenon, photosensitive rashes,
pleuritic chest pains, headaches, fatigue, fevers and lymphadenopathy are just
a few of the many non-specific presenting features of this disease.
There are no
diagnostic criteria for lupus and the ACR classification criteria are often
misused in this context and can result in missed diagnosis/under-treatment. For
example a patient may present with arthritis, Raynaud’s phenomenon,
malaise, fevers, lymphadenopathy, oral ulcers and a positive ANA. This patient
clearly may have SLE but does not fulfil the 4 criteria needed for classification
by the ACR criteria but investigation and treatment should not be delayed until
these criteria are fulfilled. The ACR criteria were specifically designed to be
highly specific for research studies to enable consistency between studies and
have been updated to include antiphospholipid
antibodies in the criteria.
The objective assessment of lupus has
depended on a number of disease activity scoring
systems which usually give a single
numeric value.
Diagnostic criteria
|
The 1982 Criteria for Classification of Systemic
Lupus Erythematosus, Updated 1997 |
||
|
1. Malar rash |
Fixed erythema, flat or raised, over the malar
eminences |
|
|
2. Discoid rash |
Erythematous raised patches with adherent keratotic
scaling and follicular plugging; atrophic scarring may occur |
|
|
3. Photosensitivity |
Exposure to UV light causes rash |
|
|
4. Oral ulcers |
Includes oral and nasopharyngeal, observed by
physician |
|
|
5. Arthritis |
Nonerosive arthritis involving two or more
peripheral joints, characterized by tenderness, swelling, or effusion |
|
|
6. Serositis |
Pleuritis or pericarditis documented by ECG or rub
or evidence of pericardial effusion |
|
|
7. Renal disorder |
Proteinuria > 0.5 g/d or > 3+, or cellular
casts |
|
|
8. Neurologic disorder |
Seizures without other cause or psychosis without
other cause |
|
|
9. Hematologic disorder |
Hemolytic anemia or leukopenia (< 4000/mL) or
lymphopenia (< 1500/mL) or thrombocytopenia (< 100,000/mL) in the
absence of offending drugs |
|
|
10. Immunologic disorder |
Anti-dsDNA, anti-Sm, and/or anti-phospholipid |
|
|
11. Antinuclear antibodies |
An abnormal titer of ANAs by immunofluorescence or
an equivalent assay at any point in time in the absence of drugs known to
induce ANAs |
|
|
If four of these criteria are present at any time
during the course of disease, a diagnosis of systemic lupus can be made with
98% specificity and 97% sensitivity. |
||
Laboratory and instrumental
investigations
Clinical examination
of all organ systems including routine urinalysis and blood pressure
measurement is mandatory. Simple investigations may yield useful information.
For example, a grossly elevated erythrocyte sedimentation rate (ESR) with a
normal C-reactive protein (CRP) is a strong pointer to lupus and related
connective tissue diseases. Blood count abnormalities such as anaemia,
neutropenia, lymphopenia and thrombocytopenia are also common.
Serologically can be found:
|
Table 1. Autoantibodies
in Patients with SLE |
|
|||
|
|
Incidence, % |
Antigen
Detected |
Clinical
Importance |
|
|
Antinuclear
antibodies |
98 |
Multiple nuclear |
Human cell substrates are more sensitive than
murine. Repeatedly negative tests make SLE unlikely. |
|
|
Anti-DNA |
70 |
DNA
(ds) |
Anti-dsDNA is relatively disease-specific;
anti-ssDNA is not. High titers are associated with nephritis and clinical
activity in some patients. |
|
|
Anti-Sm |
30 |
Protein complexed to 6 species of small nuclear RNA |
Specific for SLE. |
|
|
Anti-RNP |
40 |
Protein complexed to U1RNA |
High titer in
syndromes with features of polymyositis, lupus, scleroderma, and mixed
connective tissue disease. If present
in SLE without anti-DNA, risk for nephritis is low. |
|
|
Anti-Ro (SS-A) |
30 |
Protein
complexed to y1-y5 RNA |
Associated with
Sjogren's syndrome, subacute cutaneous lupus, inherited C¢ deficiencies,
ANA-negative lupus, lupus in the elderly, neonatal lupus, congenital heart
block. Can cause nephritis. |
|
|
Anti-La (SS-B) |
10 |
Phosphoprotein |
Always
associated with anti-Ro. Risk for nephritis is low if present. Associated
with Sjogren's syndrome. |
|
|
Antihistone |
70 |
Histones |
More frequent in
drug-induced LE (95%) than in spontaneous SLE. |
|
|
Antiphospholipid |
50 |
Phospholipids |
Three types
(lupus anticoagulant (LA), anticardiolipin (aCL), and false-positive test for
syphilis (BFP). The LA and aCL (particular high-titer IgG) are associated
with clotting, fetal loss, thrombocytopenia, and valvular heart disease.
Antibodies to b2-glycoprotein I are part of this group. |
|
|
Antierythrocyte |
60 |
Erythrocyte |
A small
proportion of these patients develop overt hemolysis. |
|
|
Antiplatelet |
30 |
Platelet surface + cytoplasm |
Associated with
thrombocytopenia in 15% of patients. |
|
|
Antilymphocyte |
70 |
Lymphocyte surface |
Probably
associated with leukopenia and abnormal T cell function. |
|
|
Antineuronal |
60 |
Neuronal and lymphocyte
surface |
In some series,
high titers of IgG correlate with diffuse CNS lupus. |
|
|
Antiribosomal P |
20 |
Ribosomal P protein |
In some series,
antibody in serum correlates with psychosis or depression due to CNS SLE. |
|
|
|
|
|
|
|
Antinuclear
antibodies are highly sensitive but not specific and anti-dsDNA antibodies are
specific but not sensitive and it is important to recognise that a negative
result for anti-dsDNA antibodies does not exclude a diagnosis of lupus.
This table is not a
standardized guideline, and tests can vary in different clinical settings. The
clinical assessment and tests must be combined to make an appropriate diagnosis
of SLE.
Diagnostic
Tests for Systemic Lupus Erythematosus
|
Test |
Possible Abnormalities |
Mechanism |
Significance and Use |
|
CBC plus differential |
Anemia, thrombocytopenia, |
Autoantibodies to |
Disease activity markers for SLE and |
|
Basic metabolic panel |
Elevated BUN/Cr ratio |
Immune complex |
Diagnosis |
|
ESR and CRP |
Elevated |
Inflammatory markers |
Disease activity marker for follow-up if |
|
Complements (C3, C4) |
Low |
Immune complex |
Disease activity marker |
|
Urine chemistry |
Proteinuria, hematuria, |
Glomerulonephritis or |
SLE nephritis and/or nephrotic syndrome |
|
LFTs |
Elevated transaminases |
Unknown |
Lupus hepatitis, nephrotic syndrome (low |
|
ANA (IFA + EIA) |
Useful as a screening test |
|
ANA-negative lupus is rare (manifests |
|
ENA panel |
Anti-Sm, anti-RNP, |
Antibodies to specific |
Anti-Sm: Highly specific for SLE |
|
dsDNA antibody |
Positive |
Antibodies to the |
Diagnostic of SLE |
|
APLAs |
Lupus anticoagulant panel, |
Antibodies to |
Moderate to high titers of IgG and IgM in |
APLA, antiphospholipid
antibody; BUN, blood urea nitrogen; CBC, complete blood count; CNS, central
nervous system; Cr, creatinine; CREST, calcinosis, Raynaud's syndrome,
esophageal involvement, sclerodactyly, telangiectasia; CRP, C-reactive protein;
CTD, connective tissue disease; dsDNA, double-stranded DNA; EIA, enzyme
immunoassay; ENA, extractable nuclear antigens; ESR, erythrocyte sedimentation
rate; IFA, immunofluorescent antibody; Ig, immunoglobulin; JIA, juvenile
idiopathic arthritis; LFTs, liver function tests; MCTD, mixed connective tissue
disease; NSAID, nonsteroidal anti-inflammatory drug; RA, rheumatoid arthritis;
RBC, red blood cell count; RNP, ribonuclear protein; SCLE, subacute cutaneous
lupus erythematosus; SLE, systemic lupus erythematosus; SS, Sjögren's
syndrome
TREATMENT
Patient Education
In order to obtain
optimal results from drug therapy, patient education plays a vital role and
must be paid due attention. Every newly diagnosed patient needs to be educated
about the disease. In this regard, pamphlets especially written for patients
can be very helpful. For illiterate patients, the treating physician or a specialist
nurse will have to spend the necessary time on education. It is often useful to
offer a new patient the opportunity to interact with other previously diagnosed
lupus patients who are identified by the specialist as having a positive
outlook of the disease and the enthusiasm to function as a counsellors. In many
advanced centres (outside India), community-based lupus support groups exist and they
perform this vital function. The biological behaviour of the disease, in
particular, the long remitting and relapsing course of the disease must be
explained to the patient. Patients also need advice regarding marriage,
contraception, pregnancy and brest-feeding. As regards marriage, the physician
should discuss the risks involved openly with the patient. Participation of the
fiancee is crucial. Avoidance
of sun-exposure Sunlight is
generally detrimental to the health of lupus patients and frequently
responsible for exacerbations of lupus activity. Photosensitive patients must
be advised to wear protective clothing with long sleeves etc, use sunscreens
(creams/lotions) with sun- protection factor (spf) of more than 15 and to avoid
going outdoors during daytime when sunlight is intense (if absolutely
necessary, they must use an umbrella to screen off sun). Sun rays reflected
from sea water around sunrise and sunset are also harmful. Infections Infections are common in SLE and
therefore, patients must get any unexplained fever evaluated promptly. This is
particularly necessary when patient is on long-term steroid/ cytotoxic therapy.
General approach to the drug
therapy of SLE
Since there is a
range of severity of disease manifestations, proper categorization based on
clinical and laboratory features is the first therapeutic step. The following
scheme is recommended:
Characterised by arthritis, arthralgia, myalgia, fatigue, mild mucocutaneous involvement, low-grade fever, mild serositis, lupus headache, musculoskeletal complaints are the commonest features of SLE. For mild symptoms, NSAIDs and analgesics may suffice. NSAIDs can occasionally cause adverse effects which may resemble those produced by the disease itself such as proteinuria, edema, renal failure and aseptic meningitis. In some patients, the above symptoms may not be alleviated with NSAIDs alone, and they should be prescribed antimalarials (chloroquine, hydroxychloroquine). These drugs are particularly useful for cutaneous manifestations of SLE. These agents have multiple properties: immunosuppressive anti-inflammatory and sun-blocking. They are also reported to possess anti-platelet and cholesterol lowering effects. The drug of choice is hydroxychloroquine (200 mg BD for 3 months and then 200 mg daily). The maintenance dose must not exceed 6 mg/kg/day. Although the incidence of retinal toxicity is very low, annual monitoring of vision with perimetery using a red object is recommended (for chloroquine, 6-monthly monitoring is desirable). The drug must be discontinued if a central scotoma is detected at any stage. Other significant side effects include nausea, pruritus, hyperpigmentation, myopathy and rarely psychosis. Use of hydroxychloroquine during pregnancy is controversial. When antimalarials are withdrawn after prolonged administration, some patients may develop a relapse of lupus activity. In refractory cases, quinacrine may be combined with hydroxychloroquine. Alternatives include dapsone and thalidomide. Quinacrine and thalidomide are, however, not available in India. Patients not responding to the above measures may be treated with low-dose steroid therapy (Prednisolone 0.3- 0.5 mg/kg/day) for 4-6 weeks followed by slow tapering. For lupus dermatitis, there is also a role of local steroids, including topical creams and ointments and injections into unresponsive skin lesions. However, the steroid cream application for facial rash is not recommended. Adequate protection against sun is essential (vide supra).
Characterised by high-grade
fever, toxaemia, severe mucocutaneous manifestations, marked photosensitivity,
moderate to severe serositis, lupus pneumonitis, mild to moderate myocarditis,
mesangioproliferative or minimal change lupus nephritis, haemolytic anaemia and
thrombocytopenia For moderate and severe manifestations, prednisolone 1 mg/kg
orally per day is the drug of choice. Antimalarials may be administered
concomitantly. High dose of steroid must be continued till disease activity is
well controlled that usually takes up to 6 weeks when it should be tapered off
slowly over 6 to12 months. In a toxic appearing patient, the administration of
intravenous pulse methylprednisolone (15 mg/kg, max. 1 g) over an hour for 3 or 5 consecutive days may achieve
rapid control of lupus activity. Dexamethasone 100 mg is a good, cheap and
equally effective alternative steroid for pulse therapy. Although rare,
arrhythmias, accelerated hypertension, psychosis, seizures and sudden death
have been reported with pulse therapy. The pulses should be followed by oral
prednisolone. Calcium
supplements (1 gm/day) and vitamin D (800 units/day) prescribed along with
steroids retard osteoporosis. Alendronate 10 mg daily or 70 mg once a week is a
good antiresorptive drug for prevention of osteoporosis in patients starting on
long-term steroid therapy. A maintenance dose of oral steroid (beyond 6 months)
is not necessary in the majority of these patients and most often it is
possible to maintain the remission with antimalarials and intermittent use of
NSAIDs. INH prophylaxis in Indian patients has been a point of debate because
of fear of promoting INH resistance.
Category III (Severe SLE)
Characterised by
organ/life-threatening features such as focal/diffuse proliferative
glomerulonephritis with or without azotaemia/hypertension, lupus cerebritis
with recurrent seizures, acute confusional state, coma; systemic necrotizing
vasculitis such as one causing peripheral gangrene, GI bleeding or mononeuritis
multiplex. A combination therapy consisting of high-dose daily oralprednisolone (40-60 mg/day) and intravenous cyclophosphamide pulses (0.75 gm/m2, maximum of 1 g, over 1 hour) is recommended. The cyclophosphamide pulses
are given once a month for 6 months by which time usually remission is achieved
and then a maintenance pulse is administered every 3 months for a total of 2
years of cytotoxic therapy. Prednisolone is tapered off or reduced to a very
low dose i.e. 5-7.5 mg per day by 6 months. At least two-thirds of patients
maintain a long-term remission after this treatment regimen. Haemorrhagic
cystitis is rare if attention is paid to adequate hydration after the pulse and
prompt voiding of bladder and co-administration of MESNA. The overall risk of
irreversible ovarian failure was noted to be 39% in one study. It was much
higher for women aged more than 30. Infertility is common in men as well and
sperm banking is recommended, if facilities are available. Bone marrow
suppression and secondary infections (Herpes zoster, tuberculosis, pneumocystis
carinii, staphylococcus, pseudomonas etc.), sclerosing cystitis and bladder
carcinoma, are the other adverse outcomes. Some authorities recommend the above
regimen for induction of remission (the first 6 months), which is then
maintained with azathioprine 2-2.5 mg/kg/day for about 2 years.
Alternatives include intravenous pulses of steroids on 3 consecutive days each
month, daily oral administration ofcyclophosphamide (2 mg/kg/day) or azathioprine from the
beginning or a combination of these two agents along with oral prednisolone.
The latter is believed to be the most potent (and the most toxic) regimen.
Cyclophosphamide based regimens have been shown to be superior in achieving
renal preservation. Plasmapheresis, methotrexate, cyclosporine and
mycophenolate mofetil are other options. Progressive organ damage may still
occur over several years despite achieving good short-term remissions in these
patients.
Category IV (SLE with
miscellaneous features)
Characterised by
antiphospholipid syndrome (recurrent DVT, CVAs, recurrent foetal loss etc.),
pure membranous lupus nephritis, chronic sclerosing lupus nephritis, seizures
without other evidence of lupus activity, behavioural disorders without other
serious manifestations, resistant thrombocytopenia or haemolytic anaemia
Immunosuppressive therapy does not play any significant role in these
conditions. Treatment of antiphospholipid syndrome is described in appendix. If
seizures or psychosis occur as isolated events with no evidence of lupus
activity elsewhere in the body, only symptomatic treatment is recommended.
Steroids are not indicated. Pure membranous glomerulonephritis (WHO Class V)
may be treated initially with prednisolone 1 mg/kg/day. If there is no response
after 6 weeks (85-90% of cases), steroids may be quickly tapered off because
they are not likely to help. There is no proven role of cytotoxic drugs in the
treatment of this condition in SLE. Renal failure occurs but is less frequent
as compared with proliferative glomerulonephritis. Chronic sclerosing
glomerulonephritis is best treated with conservative therapy, dialysis and
transplantation. Immunosuppressive therapy is not beneficial. At least, 3
months of dialysis is recommended before considering renal transplant as the
outcome of the transplant is better in patients whose lupus disease activity
remains clinically stable on dialysis for at least 3 months. For refractory
thrombocytopenia, danazol may be useful. Colchicine and vincristine are sometimes
useful to improve the platelet count. Splenectomy may be indicated in some
cases where platelet count tends to be less than 50,000/ cu mm and maintenance
requirement for steroids is high. Such patients should receive pneumococcal
vaccine. Plasmapheresis may be employed in refractory cases where steroid and
cyclophosph-amide pulses do not produce satisfactory results. Intravenous
immunoglobulin has also been used in similar situations. A few instances of
successful remission of refractory lupus following stem cell transplant are
reported.
Other specific
entities:
Transverse
myelitis : Requires aggressive treatment
with prednisolone orally 1.5 mg/kg/day and IV cyclophsophamide bolus. If there
is no improvement, plasmapheresis should be considered.
Seizures: For generalized seizure,
phenytoin and barbiturates are used and for focal, carbamazepine, valproate or
gabapentin is used.
Headaches: Most patients respond to NSAIDs. In intractable cases steroid may
be used. Chorea: No specific therapy is required.
Cranial /autonomic and peripheral neuropathy: Oral prednisolone in a dose of 1mg/kg/day is useful.
General measures: It
is advisable to restrict salt if hypertension is present, fat if hyperlipidemia
or nephrotic syndrome is present, protein should be restricted if azotaemia is
present and calcium should be supplemented with steroid therapy. Meticulous
control of hypertension is desirable. Pregnancy should be avoided during active
lupus nephritis with suitable contraception (vide infra). NSAIDs should be
avoided in the presence of impaired renal function. Immunosuppressive therapy:
This is generally guided by the WHO Class of lupus nephritis.
1. Class I: Immunosuppressive therapy is
not indicated.
2. Class IIa: Immunosuppressive therapy is
not indicated.
3. Class IIb: If proteinura is > 1
gram/24 hours, antidsDNA is high and C3 is low, prednisolone should be administered
at a dose of 20 mg daily for 6-12 weeks, followed by tapering over next 3
months.
4.Class III &
IV: Protocol
for this group is already described above (See Category III under management).
5. Class V: Described under management above (Category IV). A high chronicity index correlates with poor renal outcome with progression to end stage renal disease despite treatment. High activity Index is also associated with poor outcome if not treated aggressively with appropriate immunosuppressive therapy. Patients with high chronicity index and serum creatinine more than 3 mg/dL should not be treated aggressively unless activity index is also high. If serum creatinine is chronically high and more than 5 mg/dL, aggressive immunosuppressive therapy is harmful. Such patients will be better managed with dialysis and transplantation in due course.
1. Deep venous
thrombosis: The main purpose of treatment here is to prevent pulmonary
embolism. Standard measures include bed-rest, elevation of the affected limb to
allow the oedema and tenderness to subside and anticoagulant therapy. Heparin
and warfarin should be started simultaneously so as to allow an overlap of
about 5 days. INR should be adjusted between 3 and 4 on long-term warfarin
therapy. The duration of warfarin therapy is life-long in patients with
recurrent venous thrombosis. Thrombolytics such as streptokinase, urokinase and
tPA can be used but they are not more effective in preventing pulmonary
embolism. Thromboendarterectomy and percutaneous insertion of IVC filter may be
considered in special circumstances.
2. Acute arterial
thrombosis: In a patient with APS this usually means a TIA or stroke, with MI
and digital gangrene being less common. In some patients with acute stroke
(< 3 hours duration), thrombolytics can be used but the standard of care is
usually heparin followed by warfarin. Low-dose aspirin is strongly recommended
in patients who continue having thrombotic events despite full anticoagulation.
APS patients with acute MI can be treated with thrombolytics, angioplasty or
coronary stents. Peripheral arterial thrombosis can be treated with
thrombolytics or heparin or angioplasty.
3. Catastrophic APS:
These patients develop thrombosis in multiple organs and the features mimic DIC
and TTP. Oral contraceptives and other drugs, pregnancy, infection and surgical
procedures have been identified as predisposing factors.
Crises
of SLE
Treatment of the autoimmune
crisis
High dose of glucocorticoids
including pulses therapy
A. combination "pulses" therapy -
1000 mg of methylprednisolone
+ 1000 mg of cyclophosphamide at the first day and then at the second and third
day – only 1000 mg of methylprednisolone
B. -combination of high doses of glucocoticoids and cyclosporine A (5
mg/kg per day in the course of 6 weeks)
- plasmapheresis
Treatment of the cerebral crisis
- combination
"pulses" therapy - 1000 mg of methylprednisolone
+ 1000 mg of cyclophosphamide at the first day and then at the second and third
day – only 1000 mg of methylprednisolone
- cyclophosphanum
(cyclophosphamide) intravenous 2 g once a week for 4 weeks, then 200 mg once a week for 2 – 2,5 years
- plasmapheresis
- immune globulin 0,4 g/kg
intravenous in the course of 5 days
Treatment of the hematologic
crisis
- high dose of glucocorticoids
including pulses therapy
- combination of high doses of
glucocoticoids and cyclosporine A (5 mg/kg per day in the course of 6 weeks)
- immune globulin 0,4 g/kg
intravenous in the course of 5 days
SYSTEMIC
SCLEROSIS

Systemic sclerosis (SSc) is a chronic multisystem
disorder of unknown etiology characterized clinically by thickening of the skin
caused by accumulation of connective tissue and by involvement of visceral
organs, including the gastrointestinal tract, lungs, heart, and kidneys.
Epidemiology
SSc has a worldwide
distribution and affects all races. The onset of disease is unusual in
childhood and young men. The incidence increases with age, peaking in the third
to fifth decade. Women overall are affected approximately three times as often
as men and even more often during the late childbearing years (8:1).
Etiology
Immunologic mechanisms and heredity (certain HLA subtypes) play a role in etiology. SSc-like syndromes can result from exposure to vinyl chloride, bleomycin, pentazocine, aromatic hydrocarbons, contaminated rapeseed oil, or l-tryptophan.
IMMUNOLOGIC ABNORMALITIES: Patients with scleroderma exhibit abnormalities of the
humoral and cellular immune systems. The number of circulating В lymphocytes is normal, but there is
evidence of hyperactivity, as manifested by hypergammaglobulinemia and
cryoglobulinemia. Antinuclear antibodies are common but are usually in a lower
titer than in SLE. Antibodies virtually specific for scleroderma include nucleolar autoantibodies, antibodies to ScL-70, a nonhistone nuclear protein, and anticentromere
antibodies. Antibodies against types I and IV collagen have also been
described and may be relevant to the pathogenesis of this disease.
Cellular immune derangements in
progressive systemic sclerosis include a decrease in the number of circulating
T cells, a decrease in helper T cells, and an increase in suppressor T cells.
Although functional lymphocyte studies are inconclusive, lymphocytes
frompatients with this disease are sensitized to skin extracts or collagen.
They respond to these substances by proliferating and by producing lymphokines,
which may cause chemotaxis and enhanced collagen synthesis by fibroblasts.
Other disorders associated with
autoimmune phenomena, such as thyroiditis and primary biliary cirrhosis, are
increased in incidence in patients with scleroderma.
FIBROSIS: Progressive systemic sclerosis is characterized by excessive
collagen deposition in many tissues. Although the cause remains obscure, it is
thought that this fibrosis may be due to an abnormality in fibroblast function.
Fibroblasts from patients with this disorder show increased
collagen synthesis in tissue culture, possibly because T cells
sensitized to collagen produce lymphokines.
CHROMOSOMAL
CHANGES: Almost all (96%) of patients with scleroderma
have chromosomal abnormalities, such as chromatid breaks, translocations, and
deletions. These abnormalities are acquired rather than inherited and
are associated with a "serum breaking factor." The significance of
these chromosomal abnormalities is unclear.
PATHOLOGY
The skin in scleroderma
displays early edema and then induration, with the latter characterized by the
following:
• A striking increase in collagen
fibers in the reticular dermis
• Thinning of the epidermis with
loss of rete pegs
• Atrophy of dermal appendages
• Hyalinization and obliteration
of arterioles
• Variable mononuclear
infiltrates, consisting primarily of T cells
The stage of induration may
progress to atrophy or revert to normal. Similar histologic alterations
occur in the synovium,
lungs, gastrointestinal tract, heart, and kidneys.
Classification
|
Classification
of Scleroderma |
|
Localized Scleroderma (Localized cutaneous fibrosis) |
|
· Limited or generalized morphea: Circumscribed
patches of sclerosis · Linear scleroderma: Linear lesions seen in childhood · En coup de sabre: Linear lesions of the scalp or face |
|
Systemic Scleroderma (Cutaneous and noncutaneous
involvement) |
|
· Limited cutaneous systemic sclerosis (lcSSc),
formerly called CREST syndrome (calcinosis of the digits, Raynaud's
phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias) · Diffuse cutaneous systemic sclerosis (dcSSc):
Sclerosis of proximal extremities, trunk, and face · Systemic sclerosis sine scleroderma (ssSSc): Organ
fibrosis on; no skin thickening |
CLINICAL MANIFESTATIONS
Skin
In the
skin, a thin epidermis overlies compact bundles of collagen that lie parallel
to the epidermis. Fingerlike projections of collagen extend from the dermis
into the subcutaneous tissue and bind the skin to the underlying tissue. Dermal
appendages are atrophied, and rete pegs are lost. In early stages of disease, a
mononuclear cell infiltrate of predominantly T cells surrounds small dermal
blood vessels. Increased numbers of T cells, monocytes, plasma cells, and mast
cells are found, particularly in the lower dermis of involved skin.
Gastrointestinal Tract
In the
lower two-thirds of the esophagus, the histologic findings consist of a thin
mucosa and increased collagen in the lamina propria, submucosa, and serosa. The
degree of fibrosis is less than in the skin. Atrophy of the muscularis in the
esophagus and throughout the involved portions of the gastrointestinal tract is
more prominent than the amount of fibrotic replacement of muscle. Ulceration of
the mucosa is often present and may be due to either SSc or superimposed peptic
esophagitis. Chronic esophageal reflux can lead to metaplasia of the lower
esophagus (Barrett's esophagus), which is a premalignant lesion. Striated
muscles in the upper third of the esophagus are relatively spared. Similar
changes may be found throughout the gastrointestinal tract, especially in the
second and third portions of the duodenum, in the jejunum, and in the large
intestine. Atrophy of the muscularis of the large intestine may lead to the
development of large-mouth diverticula. In the later stages of the disease, the
involved portions of the gastrointestinal tract become dilated. Infiltration of
lymphocytes and plasma cells in the lamina propria is also present.
Lung
With
pulmonary involvement, diffuse interstitial fibrosis, thickening of the
alveolar membrane, and peribronchial and pleural fibrosis are observed.
Bronchiolar epithelial proliferation accompanies the pulmonary fibrosis.
Rupture of septa produces small cysts and areas of bullous emphysema. Small
pulmonary arteries and arterioles show intimal thickening, fragmentation of the
elastica, and muscular hypertrophy; this may occur without interstitial
pulmonary fibrosis and produce pulmonary hypertension, particularly in a subset
of patients with limited cutaneous SSc.
Musculoskeletal System
The
synovium in patients with arthritis is similar to that seen in early rheumatoid
arthritis and shows edema with infiltration of lymphocytes and plasma cells. A
characteristic finding is a thick layer of fibrin overlying and within the
synovium. Later in the disease the synovium may become fibrotic. Fibrinous
deposits appear on the surfaces of tendon sheaths and in the overlying fascia
and may lead to audible creaking over moving tendons.
Histologic features of primary
myopathy consist of interstitial and perivascular lymphocytic infiltrations,
degeneration of muscle fibers, and interstitial fibrosis. Arterioles may be
thickened, and capillaries may be decreased in number. Pathologic and
electrophysiologic findings of polymyositis in proximal muscles are present in
the few patients who are considered to have the overlap syndrome of SSc and
polymyositis.
Heart
Cardiac
involvement consists of degeneration of myocardial fibers and irregular areas
of interstitial fibrosis that are most prominent around blood vessels.
Intermittent spasm of blood vessels may result in contraction band necrosis,
similar to change observed in myocardial infarction in patients with
atherosclerotic coronary artery disease. Fibrosis also involves the conduction
system, leading to atrioventricular conduction defects and arrhythmias. The
wall of smaller coronary arteries may be thickened. Fibrinous pericarditis and
pericardial effusions are found in some patients.
Kidney
Renal
involvement is found in over half the patients and consists of intimal
hyperplasia of the interlobular arteries; fibrinoid necrosis of the afferent
arterioles, including the glomerular tuft; and thickening of the glomerular
basement membrane. Small cortical infarctions and glomerulosclerosis may be
present. The renal pathologic change is often indistinguishable from that
observed in malignant hypertension. Renal vascular lesions, however, may be
present in the absence of hypertension. Immunofluorescence studies of kidney
have shown IgM, complement components, and fibrinogen in the walls of affected
vessels. Angiographic renal studies in patients with SSc may show constriction
of the intralobular arteries, a finding that simulates the vasospasm of the
digital arteries observed in Raynaud's phenomenon. Cold-induced Raynaud's
phenomenon has been shown to decrease renal blood flow.
Other Organs
Primary
liver involvement is not common. Primary biliary cirrhosis occurs in some
patients, particularly in those with the limited cutaneous form of SSc. Fibrosis
of the thyroid gland may develop in the presence or absence of autoimmune
thyroiditis.
Thickening of the periodontal
membrane with replacement of the lamina dura is demonstrated radiographically
as widening of the periodontal space and may cause gingivitis and loosening of
the teeth. The decreased oral aperture and mucosal dryness make eating and oral
hygiene difficult.

CLINICAL
MANIFESTATIONS
|
Table 6. Clinical Features of Systemic
Sclerosis |
||||
|
|
Percent of Patients with Clinical Feature
during Course of Disease |
|
||
|
Clinical Feature |
Limited |
Diffuse |
|
|
|
Raynaud's phenomenon |
95-100 |
90-95 |
|
|
|
Skin thickening |
98b |
100 |
|
|
|
Subcutaneous calcinosis |
50 |
10 |
|
|
|
Telangiectasia |
85 |
40 |
|
|
|
Arthralgias/arthritis |
40 |
70 |
|
|
|
Myopathy |
5 |
50 |
|
|
|
Esophageal dysmotility |
80 |
80 |
|
|
|
Pulmonary fibrosis |
35 |
40 |
|
|
|
Isolated pulmonary arterial hypertension |
<10 |
<1 |
|
|
|
Congestive heart failure |
<1 |
30 |
|
|
|
Renal crisis |
<1 |
15 |
|
|
|
a Limited cutaneous and diffuse cutaneous
subsets of SSc. |
||||
|
b 2% or fewer of patients have SSc sine
scleroderma. |
||||
|
|
|
|
|
|
There are two major forms of
scleroderma, localized scleroderma and systemic scleroderma (sclerosis).
Diffuse (dcSSc) and limited (lcSSc) scleroderma are the two main types of
systemic sclerosis.
Scleroderma limited to the skin
only can be subdivided into morphea and linear scleroderma.
Localized Scleroderma
The more common form of the
disease, localized scleroderma, only affects the skin without any internal
organ involvement. It often appears in the form of waxy patches (morphea) or
streaks on the skin (linear scleroderma).
Vascular abnormalities,
especially of the microvasculature are a prominent feature of SSc. The degree
and rate of skin and internal organ involvement vary among patients. Two
subsets, however, can be identified, even though there is some overlap (Table
5).
|
Subsets of Systemic Sclerosis |
|||
|
|
Diffuse |
Limiteda |
|
|
Skin involvement |
Distal and proximal extremities, face, trunk |
Distal to elbows, face |
|
|
Raynaud's phenomenon |
Onset within 1 year or at time of skin changes |
May precede skin disease by years |
|
|
Organ involvement |
Pulmonary (interstitial fibrosis); renal
(renovascular hypertensive crisis); gastrointestinal; cardiac |
Gastrointestinal; pulmonary arterial hypertension
after 10-15 years of disease in <10% of patients; biliary cirrhosis |
|
|
Nail fold capillaries |
Dilatation and dropout |
Dilatation without significant dropout |
|
|
Antinuclear antibodies |
Antitopoisomerase 1 |
Anticentromere |
|
One subset is referred to as diffuse cutaneous scleroderma and is characterized by the rapid
development of symmetric skin thickening of proximal and distal extremities,
These criteria are not,
however, applicable to clinical practice as many patients have SSc who do not
meet these criteria. Scleroderma can also occur in a localized form limited to
the skin, subcutaneous tissue, and muscle and without systemic involvement.
Localized scleroderma occurs most often in children and young women but can
affect any age group. The two localized forms are morphea, which occurs as single
or multiple plaques of skin induration, and linear scleroderma, which involves
an extremity or face. Linear scleroderma of one side of the forehead and scalp
produces a disfiguration referred to as en coup de sabre because it resembles a
wound from a sword. It may be associated with hemiatrophy of the same side of
the face.
SSc also occurs in association
with features of other connective tissue diseases. The term overlap syndrome
has been used to describe such patients. Undifferentiated connective tissue
disease has been suggested as a designation for patients who do not have
diagnostic criteria for any one connective tissue disease.
Clinical manifestations of
scleroderma. A, Generalized morphea. B, Diffuse edema of hands. C, Firm,
thickened skin. D, Flexion contractures of fingers. E, Raynaud's phenomenon
(pallor phase).
It is not uncommon for this
less severe form of scleroderma to regress or stop progressing without
treatment.
On these photo: Ischemic
digital ulcer, telangectasias on the face, dorsum of the hand mucosa, calcinosis cutis.
Systemic
Scleroderma
Systemic scleroderma always
leads to some internal organ involvement. It is further divided into two
subsets of disease, limited or diffuse. According to LeRoy and colleagues,
limited or diffuse disease is based on the extent of skin tightening. 1 In limited disease (formerly
called CREST
CREST
syndrome
|
Subsets of Systemic Sclerosis |
|
Diffuse Cutaneous Systemic Sclerosis
(dcSSc) |
|
· Onset of Raynaud's within 1 year of onset of skin
changes (puffy or hidebound) · Truncal and acral skin involvement · Presence of tendon friction rubs · Early and significant incidence of interstitial lung
disease, oliguric renal failure, diffuse gastrointestinal disease, and
myocardial involvement · Absence of anticentromere antibodies · Nailfold capillary dilation and capillary destruction · Antitopoisomerase
antibodies (30% of patients) |
|
Limited Cutaneous Systemic Sclerosis (LcSSc) |
|
· Raynaud's phenomenon for years (occasionally decades) · Skin involvement limited to hands, face, feet, and
forearms (acral) or absent · A significant late incidence of pulmonary
hypertension, with or without interstitial lung disease, trigeminal
neuralgia, skin calcifications, telangiectasias · A high incidence of anticentromere antibodies
(70%-80%) · Dilated nailfold capillary loops, usually without
capillary dropout |
Raynaud's Phenomenon SSc usually begins insidiously; the
first symptoms are frequently Raynaud's phenomenon and puffy fingers. Some 95%
of patients will experience Raynaud's phenomenon, which is defined as episodic
vasoconstriction of small arteries and arterioles of fingers, toes, and
sometimes the tip of the nose and earlobes. Episodes are brought on by cold
exposure, vibration, or emotional stress. Patients experience pallor and/or cyanosis
followed by rubor on rewarming. Pallor and/or cyanosis are usually associated
with coldness and numbness of fingers and/or toes, and rubor with pain and
tingling. Not all patients appreciate the three color phases. A history of
digit pallor appears to be the most reliable symptom for the presence of
Raynaud's phenomenon. Raynaud's phenomenon may precede skin changes by several
months or even years in those patients who subsequently develop the limited
cutaneous form of SSc. In diffuse cutaneous SSc, skin changes are seen
typically within a year of the onset of Raynaud's phenomenon. After 2 or more
years of Raynaud's phenomenon, few patients who have this as their only symptom
will subsequently develop SSc.
Raynaud's
phenomenon is characterized by blood vessel spasms in the fingers, toes, ears
or nose, usually brought on by exposure to cold. Raynaud's phenomenon and
Raynaud's disease, a similar disorder, may occur on their own or they may be
associated with autoimmune disorders such as rheumatoid arthritis, systemic
lupus erythematosus, and most frequent in scleroderma.
In
early disease, fingers and hands are swollen. Swelling may also involve
forearms, feet, lower legs, and face. However, lower extremities are relatively
spared. This edematous phase may last for a few weeks, months, or even longer.
The edema may be pitting or nonpitting and accompanied by erythema. The skin
changes begin distally in the extremities and advance proximally. The skin
gradually becomes firm, thickened, and eventually tightly bound to underlying
subcutaneous tissue (indurative phase). In patients with diffuse cutaneous
scleroderma, skin changes will become generalized, involving initially the
extremities, followed by the face and trunk over a period of time, varying from
months to a few years. In some patients, the skin changes may develop gradually
over several years. Rapid progression of these changes over a 1- to 3-year
period is associated with a greater risk of visceral disease, particularly of
the lungs, heart, or kidneys. Also in diffuse cutaneous SSc, the skin changes
usually peak in 3 to 5 years and then slowly improve. On the other hand,
patients with limited cutaneous scleroderma will usually have a more gradual
progression of skin changes, which are restricted to fingers or distal
extremity and face and may continue to worsen. In both subsets of SSc, skin
thickening is usually greater in the distal extremity. After many years of
disease, the skin may soften and return to normal thickness or become thin and
atrophic.
In some patients, particularly
those with the limited cutaneous form of disease, calcific deposits develop in
intracutaneous and subcutaneous tissue. The sites commonly involved are
periarticular tissue, digital pads, olecranon and prepatellar bursae, and skin
along the extensor surface of the forearms.
Musculoskeletal Features
More
than half the patients with SSc complain of pain, swelling, and stiffness of
the fingers and knees. A symmetric polyarthritis resembling rheumatoid
arthritis may be seen. In more advanced stages of the disease, leathery
crepitation can be palpated over moving joints, especially the knee. Extensive
fibrotic thickening of the tendon sheaths in the wrist can produce a carpal
tunnel syndrome. Muscle weakness is usually present in patients with severe
skin involvement and, in most cases, is due to disuse atrophy. There is a
distinctive histologic myopathy that accompanies SSc that is not associated
with muscle enzyme abnormalities. A few patients develop a myositis
characterized by proximal muscle weakness and muscle enzyme elevations that are
identical to polymyositis (overlap syndrome). In addition to terminal
phalanges, resorption of bone may involve ribs, clavicle, and angle of mandible.
Hypomotility of the small
intestine produces symptoms of bloating and abdominal pain and may suggest an
intestinal obstruction or paralytic ileus (pseudoobstruction). Malabsorption
syndrome with weight loss, diarrhea, and anemia is due to bacterial overgrowth
in the atonic intestine or possibly to obliteration of lymphatics by fibrosis.
Roentgenographic features of the second and third portions of the duodenum and
of the jejunum include dilatation, loss of the usual feathery pattern, and
delayed disappearance of barium. Pneumatosis intestinalis occasionally occurs
and appears as radiolucent cysts or linear streaks within the wall of the small
intestine. Benign pneumoperitoneum may result from the rupture of these cysts.
Involvement of the large intestine may cause chronic constipation and fecal
impaction with episodes of bowel obstruction. A segment of atonic bowel may act
as a fulcrum for intussuception to occur. Barium studies of the large intestine
may show dilatation, atony, and large-mouth diverticula. Laxity of the anal
sphincter may cause incontinence or rarely anal prolapse. Some patients may
have gastrointestinal features of SSc with little or no cutaneous or other
organ involvement, referred to as SSc sine scleroderma. Vascular ectasia may
develop in the stomach and intestine and can be the source of gastrointestinal
bleeding. These dilated submucosal capillaries in the stomach appear on
endoscopy as broad stripesѕhence
the term "watermelon stomach."
Pulmonary
Features Pulmonary involvement occurs in at
least two-thirds of SSc patients and is now the leading cause of death in SSc,
replacing renal disease, which can usually be treated effectively. The most
common symptom is exertional dyspnea, often accompanied by a dry, nonproductive
cough. Bilateral basilar rales may be present. In the majority of patients,
symptoms usually correlate with radiologic evidence of pulmonary fibrosis and
with restrictive lung disease on pulmonary function tests.
Pulmonary function tests are
frequently abnormal and show a reduction in vital capacity and decreased lung
compliance. Impairment of gas exchange is reflected by a low diffusing capacity
and low PO2 with exercise. These abnormalities may be present even when the
chest radiograph is normal. Chest film may show a pattern of linear densities,
mottling, and honeycombing involving most prominently the lower two-thirds of
the lung. Early interstitial pulmonary disease can be detected by
high-resolution computed tomography (HRCT) and bronchoalveolar lavage (BAL).
Both
interstitial fibrosis and vascular lesions are found in the lungs of patients
with SSc.
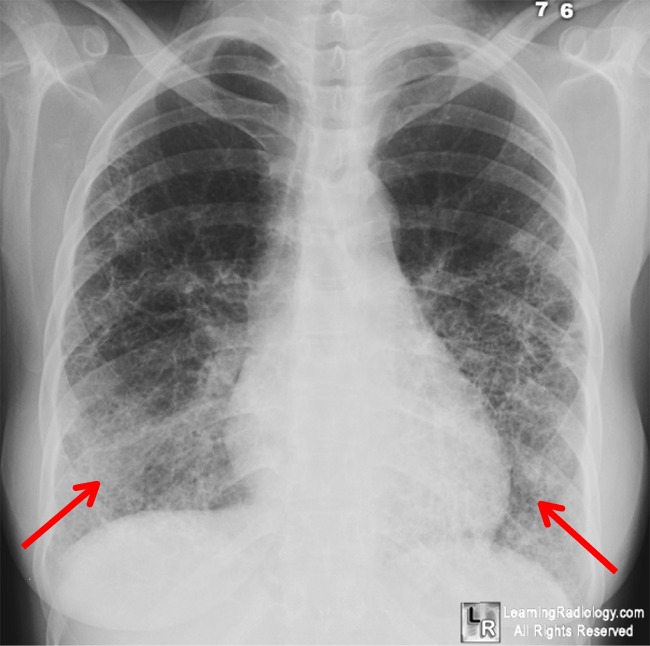{kind=link}
Interstitial
pulmonary fibrosis may be the predominant lesion in patients with diffuse or
limited cutaneous SSc. Patients with diffuse cutaneous involvement who have
antitopoisomerase 1 antibodies are particularly at risk of developing severe
pulmonary fibrosis. In the absence of significant interstitial fibrosis, a
severe form of pulmonary arterial hypertension may develop after many years of
disease in a subset of patients with limited cutaneous SSc. Fewer than 10% of
patients will develop this complication, which is caused by narrowing and
obliteration of pulmonary arteries and arterioles by intimal fibrosis and
medial hypertrophy. Pulmonary hypertension is manifested initially by
exertional dyspnea and eventually by the appearance of right-sided heart
failure. Pulmonary artery pressure can be measured noninvasively by
two-dimensional echocardiography. The prognosis is extremely poor with the
development of pulmonary hypertension; the mean duration of survival is
approximately 2 years.
A less common pulmonary problem
is aspiration pneumonia resulting from gastric reflux due to lower esophageal
atony. Restriction of chest movement caused by extensive fibrotic skin
involvement of the thorax rarely occurs. Superimposed bacterial or viral
infection can be a serious complication in patients with pulmonary fibrosis. An
increased frequency of alveolar cell and bronchogenic carcinoma is seen in
patients with pulmonary fibrosis.
Cardiac Features
Primary cardiac involvement in
SSc includes pericarditis with or without effusions, heart failure, and varying
degrees of heart block or arrhythmias. The majority of patients with diffuse
cutaneous SSc have cardiac abnormalities. Cardiomyopathy attributable to
myocardial fibrosis appears in <10% of patients and involves primarily those
patients with diffuse cutaneous scleroderma. Radionuclide studies have shown
abnormalities of left ventricular function due to myocardial fibrosis.
Cold-induced vasospasm of the hands produces defects in myocardial thallium
perfusion. The characteristic pathologic feature of contraction band necrosis
results from cardiac muscle damage caused by intermittent vasospasm of coronary
vessels. Patients may experience angina pectoris even though coronary
angiograms are normal. Patients can also develop left ventricular failure
secondary to systemic hypertension or cor pulmonale secondary to pulmonary
arterial hypertension.
Renal Features
Renal failure was the leading
cause of death in SSc until the advent of effective treatment. Significant
renal disease occurs mostly in those patients with diffuse cutaneous
scleroderma. A high risk of renal crisis is present in those patients who have
rapidly progressive widespread skin thickening in their first 2 to 3 years of
disease. Renal crisis is characterized by malignant hypertension, which can
progress rapidly to renal failure. These patients manifest hypertensive
encephalopathy, severe headache, retinopathy, seizures, and left ventricular
failure. Hematuria and proteinuria are followed by oliguria and renal failure.
The mechanism for the hypertensive crisis is activation of the
renin-angiotensin system. Before the advent of effective antihypertensive
drugs, the majority of these patients died within 6 months. A small number of
patients may develop renal crises in the absence of hypertension. Renal failure
can also develop insidiously later in the course of disease in the setting of
mild to moderate hypertension and proteinuria. In these patients or those with
clinically unrecognized renal disease, reduction of renal plasma flow secondary
to heart failure or volume depletion resulting from overdiuresis may
precipitate renal crisis. An indicator of impending renal failure is
microangiopathic anemia, which may occur in a normotensive patient. The
presence of a large chronic pericardial effusion may also herald subsequent renal
failure.
Other Features
Symptoms of dry eyes and/or dry
mouth are frequently present in patients with SSc. Lip biopsy may show
lymphocytic infiltration of minor salivary glands characteristic of Sjogren's
syndrome or intraglandular or periglandular fibrosis secondary to SSc.
Antibodies to SS-A (Ro) and/or SS-B (La) are found in those patients with lip
biopsies consistent with Sjogren's syndrome (overlap syndrome-SSc and Sjogren's
syndrome) and not in those with salivary gland fibrosis.
Hypothyroidism occurs in a
significant number of patients and may be associated with high levels of
antithyroid antibodies. Fibrosis of the thyroid gland may be present but also
occurs in the absence of autoimmune thyroiditis. Other manifestations of SSc
include trigeminal neuralgia and male impotence secondary to decreased penile
tumescence. These men have normal serum levels of testosterone and
gonadotropins. Pathogenesis of this abnormality has been considered to be
caused by vascular and/or autonomic nervous system abnormalities. Biliary
cirrhosis is occasionally observed in patients with limited cutaneous SSc.
The
1980 Criteria for systemic sclerosis (ACR)
Major
criterion:
Proximal scleroderma: symmetric
thickening, tightening, and induration of the skin of the fingers and/or the
skin proximal to the metacarpophalangeal or metatarsophalangeal joints. The
changes may affect the entire extremity, face, neck, and trunk (thorax and
abdomen).
Minor
criteria:
1. Sclerodactyly: preceding
skin changes limited to the finger.
2. Digital pitting scars or
loss of substance from the finger pad: depressed areas at tips of fingers or
loss of digital pad tissue as a result of ischemia.
3. Bibasilar pulmonary
fibrosis: bilateral reticular pattern of linear or lineonodular densities most
pronounced in basilar portions of the lungs on standard chest roentgenogram;
may assume appearance of diffuse mottlng or “honeycomb lung”. These
changes should not be attributable to primary lung disease.
*The patient should fulfi ll the major
criterion or two of the three minor criteria.
LABORATORY FINDINGS
The erythrocyte sedimentation
rate may be elevated. Hypoproliferative anemia related to chronic inflammation
is the most common cause of anemia in SSc. Anemia may also be caused by iron
deficiency secondary to gastrointestinal bleeding. Bacterial overgrowth due to
atony of the small bowel may lead to vitamin B12 and/or folic acid-deficiency
anemia. Microangiopathic hemolytic anemia is most often associated with renal
involvement and is caused by the presence of intravascular fibrin in renal
arterioles. Polyclonal hypergammaglobulinemia, consisting mostly of IgG, is
found in approximately half the patients. Rheumatoid factor, in low titer, is
present in 25% of patients. Cryoglobulins may be present in the serum.
Antinuclear antibodies detected by using a cultured human laryngeal carcinoma
cell line (HEp-2) substrate are present in 95% of patients (Table 7).
|
Table 7. Autoantibodies in Systemic Sclerosis |
|||
|
Autoantibody |
Clinical Association |
Percent |
|
|
Antitopoisomerase 1 |
Diffuse cutaneous SSc |
40 |
|
|
Anticentromere |
Limited cutaneous SSc |
60-80 |
|
|
Anti-RNA polymerase I, II, III |
Diffuse cutaneous SSc |
5-40 |
|
|
Anti-Th RNP |
Limited cutaneous SSc |
14 |
|
|
Anti-U1 RNP |
Limited cutaneous SSc Mixed connective tissue disease |
5-10 95-100 |
|
|
Anti-PM/Scl |
Overlap (SSc, polymyositis) |
25 |
|
|
|
|||
Antinuclear antibodies that have a high
specificity for SSc are antitopoisomerase 1 (Scl-70), antinucleolar, and
anticentromere. Antitopoisomerase 1, originally called anti-Scl-70, recognizes
the nuclear enzyme DNA topoisomerase 1, a nuclear enzyme involved in the unwinding of DNA for
replication and RNA transcription. These antibodies are found in ~20% of all
SSc patients and in ~40% of those with diffuse cutaneous SSc. Anticentromere
antibodies react with protein antigens located in the kinetochore region of
chromosomes and are present in 40 to 80% of patients with limited cutaneous
scleroderma or CREST syndrome. Anticentromere antibodies are found in only
about 2 to 5% of patients with diffuse cutaneous scleroderma and rarely in
other connective tissue diseases. They are found occasionally in patients with
only Raynaud's phenomenon and may indicate subsequent development of limited
cutaneous disease. The titers in MCTD are usually high (see below). Anti-SS-A
(Ro) and/or anti-SS-B (La) are present in those patients with overlap syndrome
of SSc and Sjogren's syndrome.
Laboratory Studies
The following findings may be found with
laboratory studies:
- Increased erythrocyte sedimentation
rate
- Thrombocytopenia
- Hypergammaglobulinemia
- Microangiopathic hemolytic anemia
- Increased creatine phosphokinase
levels in patients with muscle involvement
- Increased urea and creatinine levels
in patients with kidney involvement
- C-reactive protein: The nonspecific
inflammatory marker C-reactive protein was found elevated in about one quarter
of patients with systemic sclerosis, especially early disease, in whom it
correlated with disease activity, severity, poor pulmonary function, and
shorter survival.
Imaging Studies
Chest radiographs
may show normal findings in 5-10% of the patients, even when the patients have
respiratory tract symptoms. In approximately 30-60% of patients, fibrosis of
the basal parts of the lungs is observed. Occasionally, pictures of diffuse
ground-glass and honeycomb lung patterns are observed. In patients with
honeycomb lung patterns, changes are irreversible. These changes can be an
important feature of patient's response to treatment.
Bone radiography reveals generalized
osteopenia, which most commonly affects the hands. Intra-articular
calcifications often are observed.
High-resolution
computed tomography (HRCT) and scintigraphy reveal thickening of the alveolar
walls and intestinal tissue and honeycomb-appearing lungs.
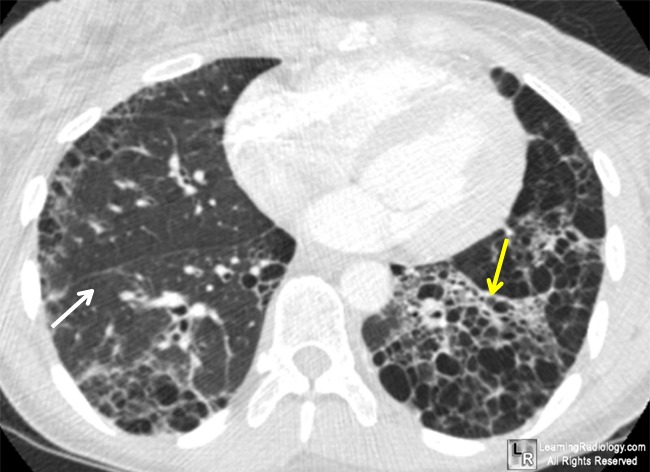{kind=link}
Gastrointestinal
tract changes may be depicted.
Scintigraphy of the esophagus may reveal a disturbance of the esophageal passage.
Cardiac and
pulmonary vascular involvement in systemic sclerosis should be evaluated.
Cardiac abnormalities may be assessed by Doppler echocardiography. Left- and right-sided heart
diseases were found to be common in persons with systemic sclerosis. A few
patients had a restrictive mitral flow pattern, possibly due to primary cardiac
involvement of systemic sclerosis.
Because cardiac
involvement is one of the major problems in systemic sclerosis, evaluation of
ventricular function using echocardiographic strain imaging should be
considered, because it appears to be useful to detect subclinical cardiac
involvement in systemic sclerosis patients with normal standard
echocardiographic and tissue Doppler velocity findings.
With bronchoalveolar lavage (BAL), abnormal numbers of granulocytes,
particularly neutrophils and eosinophils are present in BAL fluid. In addition,
the concentration of vascular endothelial growth factor may be low.
Lung function tests
reveal ventilation-perfusion changes, including the following:
- Reduced carbon monoxide diffusion
capacity
- A reduced partial pressure of oxygen,
with a normal or low partial pressure of carbon dioxide
- Restrictive ventilatory defect, with
reductions in pulmonary compliance, vital capacity, and total lung capacity
- Decreased diffusion capacity of carbon
monoxide transfer factor (DLCO) levels, which is a measure of diffusion
capacity
The gas transfer
measurement (KCO), adjusted for alveolar volume, is also reduced.
Heart changes,
including myocardial disease, pericardial problems, conduction system disease,
and arrhythmias, can be observed with the following tests:
- Electrocardiography (ECG)
- Holter 24-hour monitoring
- Doppler ultrasonography (US)
Exophthalmos,
macroglossia, and/or gigantism may be present, with increased polyphasic
potentials of normal or decreased amplitude.
Antihistone
antibodies can be observed in the course of systemic
sclerosis, but they are not characteristic. The followingantinuclear
antibodies (ANAs) are
characteristic of scleroderma:
- Antibodies against topoisomerase I DNA
(Scl 70) are detected in the serum of patients with
systemic sclerosis. The antibodies are detected in two thirds of patients
with dSSc and interstitial lung fibrosis.
- Anticentromere antibodies (ACAs) are most commonly detected in patients
with lSSc; in these patients, changes in the heart, kidneys, and lungs
(without fibrosis) are observed less frequently than in other patients.
ANAs can be detected
in the course of systemic sclerosis. ANAs include antibodies against
fibrillarin, a 34-kd protein of ribonucleoprotein U3 RNP; antibodies against
the ribonucleoprotein nucleolar 7-2 RNA protein particle Th RNP; and antibodies
to 20-110-kd proteins related to preribosomes (PM-Scl). Anti-PM/Scl antibodies
are seen in roughly 24% of patients with polymyositis/systemic sclerosis
overlap syndrome. They are also found in 3-10% of systemic sclerosis patients. The spectrum of systemic
sclerosis-associated ANA differs in patients with and without cutaneous
involvement.
Elevated
high-sensitivity C-reactive protein appears related to the occurrence of
antimitochondrial antibody in these patients.
With capillary microscopy, enlarged
capillaries are observed in all 3 portions of the capillary nail
fold–arterial, apical, and venous– and especially at the edge of
the nail fold. Adjacent areas are avascular.
Spirometry demonstrates functional lung disturbances. In approximately 70% of
patients, the DLCO is decreased.
Histologic
Findings
In the active
indurative phase, a loss of rete ridges occurs, epidermal skin appendages
atrophy, and collagen fibers in the reticular dermis appear broad and
hyalinized. A loss of space between collagen bundles is noted. Mononuclear
cells, mostly T cells, form a variable perivascular infiltrate in the deep
dermis and subcutis. Later, sclerotic changes predominate. The number of
adnexal structures is reduced, and a loss of periadnexal fat is noted.
The amount of
hyalinized collagen, myofibroblasts, mean epidermal thickness, the mononuclear
cellular infiltration, and the frequency of focal exocytosis varied
significantly between those with and those without local clinical skin
involvement.
TREATMENT
EULAR recommendations:
I. SSc-related digital vasculopathy
(Raynaud’s phenomenon, digital ulcers).
1. Nifedipine and intravenous iloprost reduce
the frequency and severity of SSc-related Raynaud’s phenomenon (SSc-RP)
attacks. Dihydropiridine-type calcium antagonists, usually oral nifedipine,
should be considered for first-line therapy for SSc-RP, and intravenous
iloprost, or other available intravenous prostanoids, should be considered for
severe SSc-RP.Intravenous prostanoids (in particular iloprost) should be
considered in the treatment of active digital ulcers in patients with SSc.
Intravenous iloprost (0.5–2 ng/kg per minute for 3–5 consecutive
days) significantly reduced the number of digital ulcers in comparison with
placebo in one small RCT.
2. Bosentan should be considered in diffuse
SSc with multiple digital ulcers, after failure of calcium antagonists and,
usually, prostanoid therapy.
II. SSc-PAH
Two high-quality RCT indicate that
bosentan improves exercise capacity, functional class and some haemodynamic
measures in pulmonary arterial hypertension (PAH). Bosentan should be strongly
considered to treat SSc-PAH. Sildenafil
(a selective type 5 phosphodiesterase inhibitor), may be considered to treat
SSc-PAH (orally at a dose of 20
mg, 40 mg or 80 mg three times a day).
III. SSc-related skin
involvement
Methotrexate may be considered
for treatment of skin manifestations of early diffuse SSc.
IV. Scleroderma interstitial
lung disease
In view of the results from two
high-quality RCT and despite its known toxicity, cyclophosphamide should be
considered for the treatment of SSc-related interstitial lung disease
(SSc-ILD). The efficacy and safety of cyclophosphamide in the treatment of
SSc-ILD was evaluated in two high. The first trial, involving 158 SSc patients
with active alveolitis, demonstrated that cyclophosphamide given orally at
a dose of 1–2 mg/kg per
day improved lung function tests, dyspnoea score and quality of life over 12
months compared.
V. Scleroderma renal crisis
Despite the lack of RCT,
experts believe that angiotensinconverting enzyme (ACE) inhibitors should be
used in the treatment of scleroderma renal crisis (SRC).
RCT evaluating the efficacy of
ACE inhibitors in the treatment of SRC are lacking. Since the first report
demonstrating a beneficial effect of ACE inhibitors in two patients with SRC,64
numerous case reports and uncontrolled studies have reported on ACE inhibitors
in SRC. A prospective analysis of 108 patients with SRC has suggested that
patients on ACE inhibitors (captopril in 47 and enalapril in eight) had a
significantly better survival rate at 1 year (76%) and 5 years (66%) compared
with patients not on ACE inhibitors (15% at 1 year and 10% at 5 years,
respectively). Treatment with ACE inhibitors was significantly associated with
better survival in
SRC, after adjustment for age
and blood pressure (p,0.001).
VI. SSc-related gastrointestinal
disease
Despite the lack of specific RCT,
experts believe that proton pump inhibitors (PPI) should be used for the
prevention of SScrelated gastro-oesophageal reflux disease (GORD), oesophageal
ulcers and strictures. Specific RCT for the efficacy of PPI in patients with
SSc are lacking. The efficacy of PPI in the treatment of GORD in a general
population is well documented in meta-analyses of RCT.
Despite the lack of specific RCT,
experts believe that prokinetic drugs should be used for the management of
SScrelated symptomatic motility disturbances (dysphagia, GORD, early response
in satiety, bloating, pseudo-obstruction, etc).
Despite the lack of
specific RCT, experts believe that, when malabsorption is caused by bacterial
overgrowth, rotating antibiotics may be useful in SSc. No RCT regarding the
efficacy of antibiotics in the treatment of SSc-related bacterial overgrowth or
malabsorption were found. In general, current treatment of small intestinal
bacterial overgrowth is based on empirical courses of broad-spectrum
antibiotics such as quinolones or amoxicillin-clavulanic acid.
References
A - Main:
1. Davidson’s Principles and practice
of medicine (21st revised ed.) / by Colledge N.R., Walker B.R., and
Ralston S.H., eds. – Churchill Livingstone, 2010. – 1376 p.
2. Harrison’s principles of internal medicine (18th edition) /
by Longo D.L., Kasper D.L., Jameson J.L. et al. (eds.). – McGraw-Hill
Professional, 2012. – 4012 p.
3. The Merck Manual of Diagnosis and Therapy
(nineteenth Edition)/ Robert Berkow, Andrew J. Fletcher and others.
– published by Merck Research Laboratories, 2011.
4. Web -sites:
A. http://intranet.tdmu.edu.ua/data/kafedra/internal/index.php?&path=vnutrmed2/classes_stud
D. http://emedicine.medscape.com/
E. http://meded.ucsd.edu/clinicalmed/introduction.htm
B - Additional:
1. Scleroderma: From Pathogenesis to
Comprehensive Management [Hardcover]/John Varga.; Christopher P. Denton.;
Fredrick M. Wigley/ Springer.- 2011.- 709 p.
2. Systemic
Lupus Erythematosus / Smolen, Josef S.;
Zielinski, Christoph C.; Geyer, G. -2012. - 200 p.
3. Clinical Rheumatology (The
Clinical Medicine Series) 12 edition/ Pacific Primary Care Software PC/ M.D.,
C. G. Weber.- 2011.- 526 p.
4. Kelley's Textbook of Rheumatology, 9th Revised edition / Firestein, Gary S.; Budd, Ralph C.; Gabriel, Sherine E.; O'Dell, James R.; McInnes, Iain B.-2012.- 2292 p.