PREPARATIONS
INFLUENCE ON THE BLOOD SYSTEM – 1: (Ferri lactas, Ferri sulfas, Ferrum Lek, Fercoven,
Hemostimulinum, Cyanocobalaminum, Acidum folicum, Pentoxilum, Natrii nucleas,
Methyluracilum, Leukogenum, Molgramostinum (leikomax), Natrii phosphas Р32)
PREPARATIONS
INFLUENCE ON THE BLOOD SYSTEM –2: (Vicasolum, Fibrinogenum, Etamzilatum, Hemostatic
sponge, Thrombinum, Calcii chloridum, Heparinum, Fraxiparine, Protamini sulfas,
Neodicumarinum, Phenilinum, Acidum acetylsalicilicum, Dipyridamolum, Ticlopidin
(Ticlid), Pentoxiphillinum)
Preparation influence on the blood system
Agents
used in Anemia’s, Hematopoietic Growth Factors
Hematopoiesis, the production from undifferentiated stem
cells of circulating erythrocytes, platelets, and leukocytes, is a remarkable
process that produces over 200 billion new cells per day in the normal person
and even greater numbers of blood cells in people with conditions that cause
loss or destruction of blood cells. The hematopoietic machinery resides
primarily in the bone marrow in adults and requires a constant supply of three
essential nutrients—iron, vitamin B12, and folic acid—as well as the presence of hematopoietic growth factors, proteins
that regulate the proliferation and differentiation of hematopoietic cells. Inadequate
supplies of either the essential nutrients or the growth factors result in
deficiency of functional blood cells.
Erythropoiesis
in bone marrow

Anemia, a
deficiency in oxygen-carrying erythrocytes, is the most common and easily treated
of these conditions, but thrombocytopenia and neutropenia are not rare and in some forms are amenable to drug therapy. In this
chapter, we first consider treatment of anemia due to deficiency of iron,
vitamin B12, or folic acid and then turn to the medical use of hematopoietic
growth factors to combat anemia, thrombocytopenia, and neutropenia.
Agents Used in Anemias
Iron
Basic Pharmacology
Iron
deficiency is the most common cause of chronic anemia—anemia that develops over
time. Like other forms of chronic anemia, iron deficiency anemia leads to
pallor, fatigue, dizziness, exertional dyspnea, and other generalized symptoms
of tissue ischemia. The cardiovascular adaptations to chronic
anemia—tachycardia, increased cardiac output, vasodilation—can worsen the
condition of patients with underlying cardiovascular disease. Iron forms the
nucleus of the iron-porphyrin heme ring, which together with globin chains
forms hemoglobin.
Hemoglobin reversibly binds oxygen and
provides the critical mechanism for oxygen delivery from the lungs to other
tissues. In the absence of adequate iron, small erythrocytes with insufficient
hemoglobin are formed, giving rise to microcytic hypochromic anemia.
Pharmacokinetics The body has an elaborate system for maintaining the supply of
the iron required for hematopoiesis.
Absorption
Iron is normally absorbed in the
duodenum and proximal jejunum, though the more distal small intestine can
absorb iron if necessary. The average diet in the USA contains 10–15 mg of
elemental iron daily.
A normal individual without iron deficiency absorbs 5–10% of this
iron, or about 0.5–1 mg daily. Iron absorption increases in response to low iron stores
or increased iron requirements. Total iron
absorption increases to 1–2 mg/d in normal menstruating women and may be as
high as 3–4 mg/d in pregnant women.
Infants and adolescents also have
increased iron requirements during rapid growth periods. Iron is available in a
wide variety of foods but is especially abundant in meat. The iron in meat
protein can be efficiently absorbed, since heme iron in meat hemoglobin and
myoglobin can be absorbed intact without first having to be broken down into
elemental iron. Iron in other foods, especially vegetables and grains, is often
tightly bound to phytates or other complexing agents and may be much less
available for absorption. Nonheme iron in foods and iron in inorganic iron
salts and complexes must be reduced to ferrous (Fe2+) iron before it can be
absorbed by the intestinal mucosal cells. Such absorption is decreased by the
presence of chelators or complexing agents in the intestinal lumen and is
increased in the presence of hydrochloric acid and vitamin C. Iron crosses the
intestinal mucosal cell by active transport. The rate of iron uptake is
regulated by mucosal cell iron stores such that more iron is transported when
stores are low. Together with iron split from absorbed heme, the newly absorbed
iron can be made available for immediate transport from the mucosal cell to the
plasma via transferrin or can be stored in the mucosal cell as ferritin, a
water-soluble complex consisting of a core crystal of ferric hydroxide covered
by a shell of a specialized storage protein called apoferritin. In general,
when total body iron stores are high and iron requirements by the body are low,
newly absorbed iron is diverted into ferritin in the intestinal mucosal cells
rather than being transported to other sites. When iron stores are low or iron
requirements are high, however, newly absorbed iron is immediately transported
from the mucosal cells to the bone marrow for the production of hemoglobin.

Iron:
possible routes of administration and fate in the organism
Transport
Iron is transported in the plasma bound to transferrin, a -globulin that
specifically binds ferric iron. The transferrin-ferric iron complex enters
maturing erythroid cells by a specific receptor mechanism. Transferrin
receptors—integral membrane glycoproteins present in large numbers on
proliferating erythroid cells—bind the transferrin-iron complex and internalize
the iron, releasing it within the cell. The transferrin and transferrin
receptor are then recycled, providing an efficient mechanism for incorporating
iron into hemoglobin in developing red blood cells. Increased erythropoiesis is
associated with an increase in the number of transferrin receptors on
developing erythroid cells. Iron store depletion and iron deficiency anemia are
associated with an increased concentration of serum transferrin.
Storage
Iron binds avidly to a protein, apoferritin, and forms the
complex ferritin. Iron is stored, primarily as ferritin, in intestinal
mucosal cells and in macrophages in the liver, spleen, and bone. Apoferritin
synthesis is regulated by the levels of free iron.
When these levels are low,
apoferritin synthesis is inhibited and the balance of iron binding shifts
toward transferrin. When free iron levels are high, more apoferritin is
produced in an effort to safely sequester more iron and protect organs from the
toxic effects of excess free iron. Ferritin is also detectable in plasma. Since
the ferritin present in plasma is in equilibrium with storage ferritin in
reticuloendothelial tissues, the plasma (or serum) ferritin level can be used
to estimate total body iron stores.
Elimination
There is no mechanism for excretion of iron. Small amounts
are lost by exfoliation of intestinal mucosal cells into the stool, and trace
amounts are excreted in bile, urine, and sweat. These losses account for no more
than 1 mg of iron per day. Because the body's ability to increase excretion of
iron is so limited, regulation of iron balance must be achieved by changing
intestinal absorption and storage of iron, in response to the body's needs.
Clinical
Pharmacology
Indications
for the Use of Iron
The only clinical indication for the use of iron preparations is the
treatment or prevention of iron deficiency anemia.
Iron deficiency is commonly seen in
populations with increased iron requirements. These include infants, especially
premature infants; children during rapid growth periods; and pregnant and
lactating women. Iron deficiency also occurs frequently after gastrectomy and
in patients with severe small bowel disease that results in generalized
malabsorption. Iron deficiency in these gastrointestinal conditions is due to
inadequate iron absorption. The most common cause of iron deficiency in adults
is blood loss. Menstruating women lose about 30 mg of iron with each menstrual
period; women with heavy menstrual bleeding may lose much more. Thus, many
premenopausal women have low iron stores or even iron deficiency. In men and
postmenopausal women, the most common site of blood loss is the
gastrointestinal tract. Patients with unexplained iron deficiency anemia should
be evaluated for occult gastrointestinal bleeding. As iron deficiency develops,
storage iron decreases and then disappears; next, serum ferritin decreases; and
then serum iron decreases and iron-binding capacity increases, resulting in a
decrease in iron-binding (transferrin) saturation.
Thereafter,
anemia begins to develop. Red cell indices
(mean corpuscular volume [MCV]: normal = 80–100 fL; mean corpuscular hemoglobin
concentration [MCHC]: normal = 32–36 g/dL) are usually low normal when iron
deficiency anemia is mild, but cells become progressively more
microcytic (low MCV) and hypochromic (low MCHC) as anemia becomes more severe.
By the time iron deficiency is diagnosed, serum iron is usually less than 40
g/dL; total iron-binding capacity (TIBC) is greater than 400 g/dL; ironbinding
saturation is less than 10%; and serum ferritin is less than 10 g/L. These
laboratory measurements can be used to confirm a diagnosis of iron deficiency
anemia in patients who present with signs and symptoms of microcytic anemia.
Treatment
The treatment of iron deficiency
anemia consists of administration of oral or parenteral iron preparations. Oral
iron corrects the anemia just as rapidly and completely as parenteral iron in most
cases if iron absorption from the gastrointestinal tract is normal.
Oral
Iron Therapy
A wide variety of oral iron preparations are available.
Since ferrous iron is most efficiently absorbed, only ferrous salts should be
used. Ferrous sulfate, ferrous gluconate, and ferrous fumarate are all
effective and inexpensive and are recommended for the treatment of most
patients. In an irondeficient individual, about 50–100 mg of iron can be
incorporated into hemoglobin daily, and about 25% of oral iron given as ferrous
salt can be absorbed. Therefore, 200–400 mg of elemental iron should be given
daily to correct iron deficiency most rapidly. Patients unable to tolerate such
large doses of iron can be given lower daily doses of iron, which results in
slower but still complete correction of iron deficiency. Treatment with oral
iron should be continued for 3–6 months. This will correct the anemia and
replenish iron stores.
Common adverse effects of oral iron therapy include nausea,
epigastric discomfort, abdominal cramps, constipation, and diarrhea. These
effects are usually dose-related and can often be overcome by lowering the
daily dose of iron or by taking the tablets immediately after or with meals.
Some patients have less severe gastrointestinal adverse effects with one iron
salt than another and benefit from changing preparations. Patients taking oral
iron develop black stools; this itself has no clinical significance but may
obscure the diagnosis of continued gastrointestinal blood loss.

Parenteral
Iron Therapy
Parenteral therapy should be reserved for patients with
documented iron deficiency unable to tolerate or absorb oral iron and patients
with extensive chronic blood loss who cannot be maintained with oral iron
alone. This includes patients with various postgastrectomy conditions and
previous small bowel resection, inflammatory bowel disease involving the
proximal small bowel, and malabsorption syndromes.
Iron dextran is a stable complex of ferric hydroxide and low-molecular-weight
dextran containing 50 mg of elemental iron per milliliter of solution. Iron-sucrose
complex and iron sodium gluconate complex are newer, alternative
preparations.
These agents can be given either by deep intramuscular
injection or by intravenous infusion. Adverse effects of parenteral iron
therapy include local pain and tissue staining (brown discoloration of the
tissues overlying the injection site), headache, light-headedness, fever,
arthralgias, nausea and vomiting, back pain, flushing, urticaria, bronchospasm,
and, rarely, anaphylaxis and death. Most adults with iron deficiency anemia
require 1–2 g of replacement iron, or 20–40 mL of iron dextran. Most physicians
prefer to give the entire dose in a single intravenous infusion in several
hundred milliliters of normal saline over 1–2 hours. Intravenous administration
eliminates the local pain and tissue staining that often occur with the
intramuscular route and allows delivery of the entire dose of iron necessary to
correct the iron deficiency at one time. There is no clear evidence that any of
the adverse effects, including anaphylaxis, are more likely to occur with
intravenous than with intramuscular administration. Owing to the risk of a
hypersensitivity reaction, a small test dose of iron dextran should always be
given before full intramuscular or intravenous doses are given. Patients with a
strong history of allergy and patients who have previously received parenteral
iron are more likely to have hypersensitivity reactions following treatment
with parenteral iron dextran.
Clinical
Toxicity
Acute Iron Toxicity
Acute iron toxicity is seen almost
exclusively in young children who have ingested a number of iron tablets.
Although adults are able to tolerate large doses of oral iron without serious consequences,
as few as ten tablets of any of the commonly available oral iron preparations
can be lethal in young children. Patients taking oral iron preparations should
be instructed to store tablets in child-proof containers out of the reach of
children. Large amounts of oral iron cause necrotizing gastroenteritis, with
vomiting, abdominal pain, and bloody diarrhea followed by shock, lethargy, and
dyspnea. Subsequently, improvement is often noted, but this may be followed by
severe metabolic acidosis, coma, and death. Urgent treatment of acute iron
toxicity is necessary, especially in young children. Activated charcoal, a
highly effective adsorbent for most toxins, does not bind iron and thus is ineffective. Whole bowel
irrigation (see Chapter 59: Management of the Poisoned Patient) should be
performed to flush out unabsorbed pills. Deferoxamine, a potent
iron-chelating compound, can be given systemically to bind iron that has
already been absorbed and to promote its excretion in urine and feces.
Appropriate supportive therapy for gastrointestinal bleeding, metabolic
acidosis, and shock must also be provided.
Chronic
Iron Toxicity
Chronic
iron toxicity (iron overload), also known as hemochromatosis, results
when excess iron is deposited in the heart, liver, pancreas, and other organs.
It can lead to organ failure and death. It most commonly
occurs in patients with inherited hemochromatosis, a disorder characterized by
excessive iron absorption, and in patients who receive many red cell
transfusions over a long period of time. Chronic iron overload in the absence
of anemia is most efficiently treated by intermittent phlebotomy. One unit of
blood can be removed every week or so until all of the excess iron is removed.
Iron chelation therapy using parenteral deferoxamine is much less efficient as
well as more complicated, expensive, and hazardous, but it can be useful for
severe iron overload that cannot be managed by phlebotomy.

VItamin
B12
Vitamin B12 serves as a cofactor for
several essential biochemical reactions in humans. Deficiency of vitamin B12
leads to anemia, gastrointestinal symptoms, and neurologic abnormalities. While
deficiency of vitamin B12 due to an inadequate supply in the diet is unusual,
deficiency of B12 in adults—especially older adults—due to abnormal absorption
of dietary vitamin B12 is a relatively common and easily treated disorder.
Chemistry
Vitamin B12 consists of a
porphyrin-like ring with a central cobalt atom attached to a nucleotide.
Various organic groups may be covalently bound to the cobalt atom, forming
different cobalamins. Deoxyadenosylcobalamin and methylcobalamin are the active
forms of the vitamin in humans.
Vitamin B12 and folate
metabolism

Cyanocobalamin and hydroxocobalamin
(both available for therapeutic use) and other cobalamins found in food
sources are converted to the above active forms. The ultimate source of vitamin
B12 is from microbial synthesis; the vitamin is not synthesized by animals or
plants. The chief dietary source of vitamin B12 is microbially derived vitamin
B12 in meat (especially liver), eggs, and dairy products. Vitamin B12 is sometimes called extrinsic
factor to differentiate it from intrinsic factor, a protein normally
secreted by the stomach.
Pharmacokinetics
The
average diet in the USA contains 5–30 g of vitamin B12 daily, 1–5 g of which is
usually absorbed.
The vitamin is avidly stored, primarily in the liver, with
an average adult having a total vitamin B12 storage pool of 3000–5000 g. Only
trace amounts of vitamin B12 are normally lost in urine and stool. Since the
normal daily requirements of vitamin B12 are only about 2 g, it would take
about 5 years for all of the stored vitamin B12 to be exhausted and for
megaloblastic anemia to develop if B12 absorption stopped.
Vitamin B12 in physiologic amounts
is absorbed only after it complexes with intrinsic factor, a
glycoprotein secreted by the parietal cells of the gastric mucosa. Intrinsic
factor combines with the vitamin B12 that is liberated from dietary sources in
the stomach and duodenum, and the intrinsic factor-vitamin B12 complex is
subsequently absorbed in the distal ileum by a highly specific
receptor-mediated transport system.
Vitamin B12 deficiency in humans
most often results from malabsorption of vitamin B12, due either to lack of
intrinsic factor or to loss or malfunction of the specific absorptive mechanism
in the distal ileum. Nutritional deficiency is rare but may be seen in strict
vegetarians after many years without meat, eggs, or dairy products. Once
absorbed, vitamin B12 is transported to the various cells of the body bound to
a plasma glycoprotein, transcobalamin II. Excess vitamin B12 is transported to
the liver for storage. Significant amounts of vitamin B12 are excreted in the
urine only when very large amounts are given parenterally, overcoming the
binding capacities of the transcobalamins (50–100 g).

Pharmacodynamics
Two essential enzymatic reactions in
humans require vitamin B12. In one, methylcobalamin serves as an intermediate in
the transfer of a methyl group from N_5-
methyltetrahydrofolate to methionine.
In the absence of vitamin B12,
conversion of the major dietary and storage folate, N5-methyltetrahydrofolate,
to tetrahydrofolate, the precursor of folate cofactors, cannot occur. As a
result, a deficiency of folate cofactors necessary for several biochemical
reactions involving the transfer of one-carbon groups develops. In particular,
the depletion of tetrahydrofolate prevents synthesis of adequate supplies of
the deoxythymidylate (dTMP) and purines required for DNA synthesis in rapidly
dividing cells.

The accumulation of folate as N5-methyltetrahydrofolate
and the associated depletion of tetrahydrofolate cofactors in vitamin B12
deficiency have been referred to as the "methylfolate trap." This is
the biochemical step whereby vitamin B12 and folic acid metabolism are linked
and explains why the megaloblastic anemia of vitamin B12 deficiency can be partially
corrected by ingestion of relatively large amounts of folic acid. Folic acid
can be reduced to dihydrofolate by the enzyme dihydrofolate reductase and thus serve as a source of the
tetrahydrofolate required for synthesis of the purines and dTMP that are needed
for DNA synthesis.
The other enzymatic reaction that
requires vitamin B12 is isomerization of methylmalonyl-CoA to succinyl-CoA by
the enzyme methylmalonyl-CoA mutase. In vitamin B12 deficiency, this conversion
cannot take place, and the substrate, methylmalonyl-CoA, accumulates.
In the past, it was thought that
abnormal accumulation of methylmalonyl-CoA causes the neurologic manifestations
of vitamin B12 deficiency. However, newer evidence instead implicates the
disruption of the methionine synthesis pathway as the cause of neurologic
problems. Whatever the biochemical explanation for neurologic damage, the
important point is that administration of folic acid in the setting of vitamin
B12 deficiency will not prevent neurologic
manifestations even though it will largely correct the anemia caused by the vitamin B12
deficiency.
Clinical Pharmacology
Vitamin B12 is used to treat or
prevent deficiency. There is no evidence that vitamin B12 injections have any
benefit in persons who do not have vitamin B12 deficiency. The most
characteristic clinical manifestation of vitamin B12 deficiency is
megaloblastic anemia. The typical clinical findings in megaloblastic anemia are
macrocytic anemia (MCV usually > 120 fL), often with associated mild or
moderate leukopenia or thrombocytopenia (or both), and a characteristic
hypercellular bone marrow with megaloblastic maturation of erythroid and other
precursor cells.
Vitamin B12 deficiency also causes a
neurologic syndrome that usually begins with paresthesias and weakness in
peripheral nerves and progresses to spasticity, ataxia, and
other central nervous system dysfunctions. A characteristic
pathologic feature of the neurologic syndrome is degeneration of myelin sheaths
followed by disruption of axons in the dorsal and lateral horns of the spinal
cord and in peripheral nerves. Correction of vitamin B12 deficiency arrests the
progression of neurologic disease, but it may not fully reverse neurologic
symptoms that have been present for several months.
Although most patients with neurologic abnormalities caused
by vitamin B12 deficiency have full-blown megaloblastic anemias when first
seen, occasional patients have few if any hematologic abnormalities. Once a
diagnosis of megaloblastic anemia is made, it must be determined whether
vitamin B12 or folic acid deficiency is the cause. (Other causes of
megaloblastic anemia are very rare.)

This can usually be accomplished by
measuring serum levels of the vitamins. The Schilling test, which measures
absorption and urinary excretion of radioactively labeled vitamin B12, can be
used to further define the mechanism of vitamin B12 malabsorption when this is
found to be the cause of the megaloblastic anemia.
The most common causes of vitamin
B12 deficiency are pernicious anemia, partial or total gastrectomy, and
diseases that affect the distal ileum, such as malabsorption syndromes,
inflammatory bowel disease, or small bowel resection.
Pernicious anemia results from defective secretion of
intrinsic factor by the gastric mucosal cells.
Patients with pernicious anemia have
gastric atrophy and fail to secrete intrinsic factor (as well as hydrochloric
acid). The Schilling test shows diminished absorption of radioactively labeled
vitamin B12, which is corrected when hog intrinsic factor is administered with
radioactive B12, since the vitamin can then be normally absorbed.
Vitamin B12 deficiency also occurs
when the region of the distal ileum that absorbs the vitamin B12-intrinsic
factor complex is damaged, as when the ileum is involved with inflammatory
bowel disease, or when the ileum is surgically resected. In these situations,
radioactively labeled vitamin B12 is not absorbed in the Schilling test, even
when intrinsic factor is added. Other rare causes of vitamin B12 deficiency
include bacterial overgrowth of the small bowel, chronic pancreatitis, and
thyroid disease. Rare cases of vitamin B12 deficiency in children have been
found to be secondary to congenital deficiency of intrinsic factor and
congenital selective vitamin B12 malabsorption due to defects of the receptor
sites in the distal ileum. Since almost all cases of vitamin B12 deficiency are
caused by malabsorption of the vitamin, parenteral injections of vitamin B12
are required for therapy. For patients with potentially reversible diseases,
the underlying disease should be treated after initial treatment with
parenteral vitamin B12. Most patients, however, do not have curable deficiency
syndromes and require lifelong treatment with vitamin B12 injections.
Vitamin B12 for parenteral injection
is available as cyanocobalamin or hydroxocobalamin. Hydroxocobalamin is
preferred because it is more highly protein-bound and therefore remains longer
in the circulation. Initial therapy should consist of 100–1000 g of vitamin B12
intramuscularly daily or every other day for 1–2 weeks to replenish body
stores.
Maintenance therapy consists of
100–1000 g intramuscularly once a month for life. If neurologic abnormalities
are present, maintenance therapy injections should be given every 1–2 weeks for
6 months before switching to monthly injections.
Oral vitamin B12-intrinsic factor
mixtures and liver extracts should not be used to treat vitamin B12 deficiency;
however, oral doses of 1000 g of vitamin B12 daily are usually sufficient to
treat patients with pernicious anemia who refuse or cannot tolerate the
injections.
Folic
Acid
Reduced forms of folic acid are
required for essential biochemical reactions that provide precursors for the synthesis
of amino acids, purines, and DNA. Folate deficiency is not uncommon, even
though the deficiency is easily corrected by administration of folic acid. The
consequences of folate deficiency go beyond the problem of anemia because
folate deficiency is implicated as a cause of congenital malformations in
newborns and may play a role in vascular disease (see Folic Acid
Supplementation: A Public Health Dilemma).

Chemistry
Folic acid (pteroylglutamic acid) is
a compound composed of a heterocycle, p-aminobenzoic acid, and glutamic
acid. Various numbers of glutamic acid moieties may be attached to the pteroyl
portion of the molecule, resulting in monoglutamates, triglutamates, or
polyglutamates. Folic acid can undergo
reduction, catalyzed by the enzyme dihydrofolate reductase ("folate
reductase"), to give dihydrofolic acid. Tetrahydrofolate can subsequently
be transformed to folate cofactors possessing one-carbon units attached to the
5-nitrogen, to the 10- nitrogen, or to both positions. The folate cofactors are
interconvertible by various enzymatic reactions and serve the important
biochemical function of donating one-carbon units at various levels of
oxidation. In most of these, tetrahydrofolate is regenerated and becomes
available for reutilization.
Pharmacokinetics
The average diet in the USA contains
500–700 g of folates daily, 50–200 g of which is usually absorbed, depending on
metabolic requirements (pregnant women may absorb as much as 300–400 g of folic
acid daily). Various forms of folic acid are present in a wide variety of plant
and animal tissues; the richest sources are yeast, liver, kidney, and green
vegetables. Normally, 5–20 mg of folates are stored in the liver and other
tissues.
Folates are excreted in the urine
and stool and are also destroyed by catabolism, so serum levels fall within a
few days when intake is diminished. Since body stores of folates are relatively
low and daily requirements high, folic acid deficiency and megaloblastic anemia
can develop within 1–6 months after the intake of folic acid stops, depending
on the patient's nutritional status and the rate of folate utilization.
Unaltered folic acid is readily and completely absorbed in the proximal
jejunum. Dietary folates, however, consist primarily of polyglutamate forms of N_5-methyltetrahydrofolate.
Before absorption, all but one of the glutamyl residues of
the polyglutamates must be hydrolyzed by the enzyme -1-glutamyl transferase
("conjugase") within the brush border of the intestinal mucosa The monoglutamate N_5-methyltetrahydrofolate
is subsequently transported into the bloodstream by both active and passive
transport and is then widely distributed throughout the body. Inside cells, N_5-methyltetrahydrofolate
is converted to tetrahydrofolate by the demethylation reaction that requires
vitamin B12.
Pharmacodynamics
Tetrahydrofolate cofactors
participate in one-carbon transfer reactions. As described above in the section
on vitamin B12, one of these essential reactions produces the dTMP needed for DNA
synthesis. In this reaction, the enzyme thymidylate synthase catalyzes the
transfer of the one-carbon unit of N_5,N_10-methylenetetrahydrofolate
to deoxyuridine monophosphate (dUMP) to form dTMP. Unlike all of the other
enzymatic reactions that utilize folate cofactors, in this reaction the
cofactor is oxidized to dihydrofolate, and for each mole of dTMP produced, one
mole of tetrahydrofolate is consumed. In rapidly proliferating tissues,
considerable amounts of tetrahydrofolate can be consumed in this reaction, and
continued DNA synthesis requires continued regeneration of tetrahydrofolate by
reduction of dihydrofolate, catalyzed by the enzyme dihydrofolate reductase.
The tetrahydrofolate thus produced can then reform the cofactor N_5,N_10-methylenetetrahydrofolate
by the action of serine transhydroxy- methylase and thus allow for the
continued synthesis of dTMP. The combined catalytic activities of dTMP
synthase, dihydrofolate reductase, and serine transhydroxymethylase are often
referred to as the dTMP synthesis cycle. Enzymes in the dTMP cycle are the
targets of two anticancer drugs; methotrexate inhibits dihydrofolate reductase,
and a metabolite of 5-fluorouracil inhibits thymidylate synthase. Cofactors of
tetrahydrofolate participate in several other essential reactions. As described
above, N_5-methy- lenetetrahydrofolate is required for the vitamin
B12-dependent reaction that generates methionine from homocysteine. In
addition,
tetrahydrofolate cofactors donate one-carbon units during
the de novo synthesis of essential purines.
In these reactions, tetrahydrofolate
is regenerated and can reenter the tetrahydrofolate cofactor pool.
Clinical
Pharmacology
Folate deficiency results in a
megaloblastic anemia that is microscopically indistinguishable from the anemia
caused by vitamin B12 deficiency (see above). However, folate deficiency does
not cause the characteristic neurologic syndrome seen in vitamin B12
deficiency. In patients with megaloblastic anemia, folate status is assessed
with assays for serum folate or for red blood cell folate.
Red blood cell folate levels are
often of greater diagnostic value than serum levels, since serum folate levels
tend to be quite labile and do not necessarily reflect tissue levels. Folic
acid deficiency, unlike vitamin B12 deficiency, is often caused by inadequate
dietary intake of folates. Alcoholics and patients with liver disease develop
folic acid deficiency because of poor diet and diminished hepatic storage of
folates. There is also evidence that alcohol and liver disease interfere with
absorption and metabolism of folates. Pregnant women and patients with
hemolytic anemia have increased folate requirements and may become folic
acid-deficient, especially if their diets are marginal. Evidence implicates
maternal folic acid deficiency in the occurrence of fetal neural tube defects,
eg, spina bifida.
Patients with malabsorption
syndromes also frequently develop folic acid deficiency. Folic acid deficiency
is occasionally associated with cancer, leukemia, myeloproliferative disorders,
certain chronic skin disorders, and other chronic debilitating diseases.
Patients who require renal dialysis also develop folic acid deficiency, because
folates are removed from the plasma each time the patient is dialyzed. Folic
acid deficiency can be caused by drugs that interfere with folate absorption or
metabolism. Phenytoin, some other anticonvulsants, oral contraceptives, and
isoniazid can cause folic acid deficiency by interfering with folic acid
absorption. Other drugs such as methotrexate and, to a lesser extent,
trimethoprim and pyrimethamine, inhibit dihydrofolate reductase and may result
in a deficiency of folate cofactors and ultimately in megaloblastic anemia.
Parenteral administration of folic
acid is rarely necessary, since oral folic acid is well absorbed even in
patients with malabsorption syndromes.
A dose of
1 mg of folic acid orally daily is sufficient to reverse megaloblastic anemia,
restore normal serum folate levels, and replenish body stores of folates in
almost all patients.
Therapy should be continued until
the underlying cause of the deficiency is removed or corrected. Therapy may be
required indefinitely for patients with malabsorption or dietary inadequacy.
Folic acid supplementation to
prevent folic acid deficiency should be considered in high-risk patients,
including pregnant women, alcoholics, and patients with hemolytic anemia, liver
disease, certain skin diseases, and patients on renal dialysis.
By January 1998, all products made
from enriched grains in the USA were required to be supplemented with folic
acid. This FDA ruling was issued to reduce the incidence of congenital neural
tube defects. Scientific studies show a strong correlation between maternal
folic acid deficiency and the incidence of neural tube defects such as spinal
bifida and anencephaly. The FDA requirement for folic acid supplementation is a
public health measure aimed at the significant number of women in the USA who
do not receive prenatal care and are not aware of the importance of adequate
folic acid ingestion for preventing birth defects in their babies. Pregnant
women have increased requirements for folic acid; at least 400 g/d is
recommended. It is estimated that the level of folic acid fortification now
required in enriched grain products provides an additional 80–100 g of folic
acid per day to the diet of women of childbearing age and 70–120 g/d to the
diet of middle-aged and older adults. There may be an added benefit for adults.
N_5-methyltetrahydrofolate is required for the conversion of
homocysteine to methionine. Impaired synthesis of N_5-methyltetrahydrofolate
results in elevated serum concentrations of homocysteine. Data from several
sources suggest a positive correlation between elevated serum homocysteine and
occlusive vascular diseases such as ischemic heart disease and stroke.
Clinical data suggest that the
folate supplementation program has improved the folate status and reduced the
prevalence of hyperhomocysteinemia in a population of middle-aged and older
adults who did not use vitamin supplements. It is possible, though as yet
unproved, that the increased ingestion of folic acid will also reduce the risk
of vascular disease in this population. While these two potential benefits of
supplemental folic acid are compelling, the decision to require folic acid in
grains was—and still is—controversial. As described in the text, ingestion of
folic acid can partially or totally correct the anemia caused by vitamin B12
deficiency. However, folic acid supplementation will not prevent the potentially irreversible neurologic damage
caused by vitamin B12 deficiency. People with pernicious anemia and other forms
of vitamin B12 deficiency are usually identified because of signs and symptoms
of anemia, which tend to occur before neurologic symptoms. The opponents of
folic acid supplementation are concerned that increased folic acid intake in
the general population will mask vitamin B12 deficiency and increase the
prevalence of neurologic disease in our elderly population. To put this in
perspective, approximately 4000 pregnancies, including 2500 live births, in the
USA each year are affected by neural tube defects. In contrast, it is estimated
that over 10% of the elderly population in the USA, or several million people,
are at risk of the neuropsychiatric complications of vitamin B12 deficiency
(Rothenberg, 1999). In acknowledgment of this controversy, the FDA kept its
requirements for folic acid supplementation at a somewhat low level. They also
recommend that all adults should keep their ingestion of folic acid below 1
mg/d.
Hematopoietic Growth
Factors
The hematopoietic growth factors are
glycoprotein hormones that regulate the proliferation and differentiation of
hematopoietic progenitor cells in the bone marrow. The first growth factors to
be identified were called colony-stimulating factors because they could
stimulate the growth of colonies of various bone marrow progenitor cells in
vitro. In the past decade, many of these growth factors have been purified and
cloned, and their effects on hematopoiesis have been extensively studied.
Quantities of these growth factors sufficient for clinical use are produced by
recombinant DNA technology.
Of the known hematopoietic growth factors, erythropoietin (epoetin
alfa), granulocyte colonystimulating factor (G-CSF), granulocyte-macrophage
colony-stimulating factor (GM-CSF), and interleukin 11 are currently
in clinical use.
Thrombopoietin is undergoing
clinical trials and will probably become available soon. Other potentially useful
hematopoietic factors are still indevelopment.
The hematopoietic growth factors
have complex effects on the function of a wide variety of cell types, including
nonhematologic cells. Their utility in other areas of medicine, particularly as
potential anticancer and anti-inflammatory drugs, is being investigated.
Erythropoietin
Chemistry & Pharmacokinetics
Erythropoietin,
a 34-39 kDA glycoprotein, was the first human hematopoietic growth factor to be
isolated. It was originally purified from the urine of patients with severe
anemia. Recombinant human erythropoietin (rHuEpo, epoetin alfa) is produced in
a mammalian cell expression system using recombinant DNA technology. After
intravenous administration, erythropoietin has a serum half-life of 4–13 hours
in patients with chronic renal failure. It is not cleared by dialysis. It is
measured in international units (IU). Darbopoetin alfa is a glycosylated form
of erythropoietin and differs from it functionally only in having a twofold to
threefold longer half-life.
Pharmacodynamics
Erythropoietin stimulates erythroid
proliferation and differentiation by interacting with specific erythropoietin
receptors on red cell progenitors. It also induces release of reticulocytes
from the bone marrow. Endogenous erythropoietin is produced by the kidney in
response to tissue hypoxia. When anemia occurs, more erythropoietin is produced
by the kidney, signaling the bone marrow to produce more red blood cells. This
results in correction of the anemia provided that bone marrow response is not
impaired by red cell nutritional deficiency (especially iron deficiency),
primary bone marrow disorders (see below), or bone marrow suppression from
drugs or chronic diseases.
Normally there is an inverse
relationship between the hematocrit or hemoglobin level and the serum
erythropoietin level. Nonanemic individuals have serum erythropoietin levels of
less than 20 IU/L. As the hematocrit and hemoglobin levels fall and anemia
becomes more severe, the serum erythropoietin level rises exponentially.
Patients with moderately severe anemias usually have erythropoietin levels in
the 100–500 IU/L range, and patients with severe anemias may have levels of
thousands of IU/L. The most important exception to this inverse relationship is
in the anemia of chronic renal failure. In patients with renal disease,
erythropoietin levels are usually low because the kidneys cannot produce the
growth factor. These patients are the most likely to respond to treatment with
exogenous erythropoietin. In most primary bone marrow disorders (aplastic
anemia, leukemias, myeloproliferative and myelodysplastic disorders, etc) and
most nutritional and secondary anemias, endogenous erythropoietin levels are
high, so there is less likelihood of a response to exogenous erythropoietin
(but see below).
Clinical
Pharmacology
The availability of erythropoietin
has had a significant positive impact for patients with chronic renal failure.
Erythropoietin consistently improves the hematocrit and hemoglobin level and
usually eliminates the need for transfusions in these patients. An increase in
reticulocyte count is usually observed in about 10 days and an increase in
hematocrit and hemoglobin levels in 2–6 weeks. Most patients can maintain a
hematocrit of about 35% with erythropoietin doses of 50–150 IU/kg intravenously
or subcutaneously three times a week. Failure to respond to erythropoietin is
most commonly due to concurrent iron deficiency, which can be corrected by
giving oral iron. Folate supplementation may also be necessary in some
patients.
In selected patients, erythropoietin
may also be useful for the treatment of anemia due to primary bone marrow
disorders and secondary anemias. This includes patients with aplastic anemia
and other bone marrow failure states, myeloproliferative and myelodysplastic
disorders, multiple myeloma and perhaps other chronic bone marrow malignancies,
and the anemias associated with chronic inflammation, AIDS, and cancer.
Patients with these disorders who have disproportionately low serum
erythropoietin levels for their degree of anemia are most likely to respond to
treatment with this growth factor. Patients with endogenous erythropoietin
levels of less than 100 IU/L have the best chance of response, though patients
with erythropoietin levels between 100 and 500 IU/L respond occasionally. These
patients generally require higher erythropoietin doses (150–300 IU/kg three
times a week) to achieve a response, and responses are often incomplete.
Erythropoietin has been used
successfully to offset the anemia produced by zidovudine treatment in patients
with HIV infection and in the treatment of the anemia of prematurity. It can
also be used to accelerate erythropoiesis after phlebotomies, when blood is
being collected for autologous transfusion for elective surgery, or for
treatment of iron overload (hemochromatosis).
Erythropoietin is one of the drugs
banned by the International Olympic Committee. The use of erythropoietin by
athletes is based on their hope that increased red blood cell concentration
will increase oxygen delivery and improve performance.
Toxicity
The most common adverse effects of erythropoietin are
associated with a rapid increase in hematocrit and hemoglobin and include hypertension
and thrombotic complications. These difficulties can be minimized by raising
the hematocrit and hemoglobin slowly and by adequately monitoring and treating
hypertension. Allergic reactions have been infrequent and mild.

Myeloid
Growth Factors
Chemistry
& Pharmacokinetics
G-CSF and GM-CSF,
the two myeloid growth factors currently available for clinical use, were
originally purified from cultured human cell lines. Recombinant human G-CSF (rHuG-CSF;
filgrastim) is produced in a bacterial expression system using recombinant
DNA technology. It is a nonglycosylated peptide of 175 amino acids, with a
molecular weight of 18 kDa. Recombinant human GM-CSF (rHuGM-CSF;
sargramostim) is produced in a yeast expression system using recombinant
DNA technology. It is a partially glycosylated peptide of 127 amino acids, with
three molecular species with molecular weights of 15,500, 15,800, and 19,500.
These preparations have serum half-lives of 2–7 hours after intravenous or subcutaneous
administration. Pegfilgrastim, a covalent conjugation product of
filgrastim and a form of polyethylene glycol, has a much longer serum half-life
than recombinant G-CSF, and so it can be injected once per myelosuppressive
chemotherapy cycle instead of daily for several days.
Pharmacodynamics
The myeloid growth factors stimulate
proliferation and differentiation by interacting with specific receptors found
on various myeloid progenitor cells. These receptors are members of the
superfamily of receptors that transduce signals by association with cytoplasmic
tyrosine kinases in the JAK/STAT pathway. G-CSF stimulates proliferation and
differentiation of progenitors already committed to the neutrophil lineage. It
also activates the phagocytic activity of mature neutrophils and prolongs their
survival in the circulation. G-CSF also has a remarkable ability to mobilize
hematopoietic stem cells, ie, to increase their concentration in peripheral
blood. This biologic effect underlies a major advance in transplantation—the
use of peripheral blood stem cells (PBSCs) instead of bone marrow stem cells
for autologous and allogeneic hematopoietic stem cell transplantation (see
below).
GM-CSF has broader biologic actions than G-CSF. It is a
multipotential hematopoietic growth factor that stimulates proliferation and
differentiation of early and late granulocytic progenitor cells as well as
erythroid and megakaryocyte progenitors. Like G-CSF, GM-CSF also stimulates the
function of mature neutrophils. GM-CSF acts together with interleukin-2 to
stimulate T cell proliferation and appears to be a locally active factor at the
site of inflammation. GM-CSF mobilizes peripheral blood stem cells, but it is
significantly less efficacious than G-CSF in this regard.
Clinical
Pharmacology
Neutropenia, a common adverse effect
of the cytotoxic drugs used to treat cancer, puts patients at high risk of
serious infection. Unlike the treatment of anemia and thrombocytopenia,
transfusion of neutropenic patients with granulocytes collected from donors is
performed rarely and with limited success. The introduction of G-CSF in 1991
represented a milestone in the treatment of chemotherapy-induced neutropenia.
This growth factor dramatically accelerates the rate of neutrophil recovery
after dose-intensive myelosuppressive chemotherapy. It reduces the duration of
neutropenia and usually raises the nadir, the lowest neutrophil count seen
following a cycle of chemotherapy.
While the ability of G-CSF to
increase neutrophil counts after myelosuppressive chemotherapy is nearly
universal, its impact upon clinical outcomes is more variable. Some clinical
trials have shown that G-CSF reduces episodes of febrile neutropenia,
requirements for broad-spectrum antibiotics, and days of hospitalization;
however, other trials failed to find these favorable outcomes. To date, no
clinical trial has shown improved survival in cancer patients treated with
GCSF. Clinical guidelines for the use of G-CSF after cytotoxic chemotherapy
have been published (Ozer et al, 2001). These guidelines recommend reserving
G-CSF for patients with a prior episode of febrile neutropenia after cytotoxic
chemotherapy, patients receiving dose-intensive chemotherapy, patients at high
risk of febrile neutropenia, and patients who are unlikely to survive an
episode of febrile neutropenia. Pegfilgrastim is an alternative to G-CSF for
prevention of chemotherapy-induced febrile neutropenia. Pegfilgrastim can be
administered less frequently, and it may shorten the period of severe
neutropenia slightly more than G-CSF. Like G-CSF and pegfilgrastim, GM-CSF also
reduces the duration of neutropenia after cytotoxic chemotherapy. It has been
more difficult to show that GM-CSF reduces the incidence of febrile
neutropenia, probably because GM-CSF itself can induce fever. In the treatment
of chemotherapyinduced neutropenia, G-CSF, 5 g/kg/d, or GM-CSF, 250 g/m2/d, is
usually started within 24–72 hours after completing chemotherapy and is
continued until the absolute neutrophil count is >10,000 cells/ L. Pegfilgrastim
is given as a single dose instead of as daily injections.
The utility and safety of the
myeloid growth factors in the postchemotherapy supportive care of patients with
acute myeloid leukemia (AML) has been the subject of a number of clinical
trials. Since leukemic cells arise from progenitors whose proliferation and
differentiation are normally regulated by hematopoietic growth factors,
including GM-CSF and G-CSF, there was concern that myeloid growth factors could
stimulate leukemic cell growth and increase the rate of relapse. The results of
randomized clinical trials suggest that both G-CSF and GM-CSF are safe
following induction and consolidation treatment of myeloid and lymphoblastic
leukemia.
There has been no evidence that
these growth factors reduce the rate of remission or increase relapse rate. On
the contrary, the growth factors accelerate neutrophil recovery and reduce
infection rates and days of hospitalization. Both G-CSF and GM-CSF have FDA
approval for treatment of patients with AML.
G-CSF and GM-CSF have also been
shown to be effective in treating the neutropenia associated with congenital
neutropenia, cyclic neutropenia, myelodysplasia, and aplastic anemia. Many
patients with these disorders respond with a prompt and sometimes dramatic increase
in neutrophil count. In some cases this results in a decrease in the frequency
of infections. Since G-CSF and GMCSF do not stimulate the formation of
erythrocytes or platelets, they are sometimes used in combination with other
growth factors for treatment of pancytopenia.
The
myeloid growth factors play an important role in autologous stem cell
transplantation for patients undergoing high-dose chemotherapy. High-dose
chemotherapy with autologous stem cell support is increasingly being used to
treat patients with tumors that are resistant to standard doses of
chemotherapeutic drugs. The high-dose regimens produce extreme
myelosuppression; the myelosuppression is then counteracted by reinfusion of
the patient's own hematopoietic stem cells (which are collected prior to
chemotherapy). The administration of G-CSF or GM-CSF early after autologous
stem cell transplantation has been shown to reduce the time to engraftment and
to recovery from neutropenia in patients receiving stem cells obtained either
from bone marrow or from peripheral blood. These effects are seen in patients
being treated for lymphoma or for solid tumors. G-CSF and GM-CSF are also used
to support patients who have received allogeneic bone marrow transplantation
for treatment of hematologic malignancies or bone marrow failure states. In
this setting, the growth factors speed the recovery from neutropenia without
increasing the incidence of acute graft-versus-host disease.
Probably the most important role of
the myeloid growth factors in transplantation is for mobilization of peripheral
blood stem cells (PBSCs). Stem cells collected from peripheral blood have
nearly replaced bone marrow as the hematopoietic preparation used for
autologous transplantation. The cells can be collected in an outpatient setting
with a procedure that avoids much of the risk and discomfort of bone marrow
collection, including the need for general anesthesia. In addition, there is
evidence that PBSC transplantation results in more rapid engraftment of all
hematopoietic cell lineages and in reduced rates of graft failure or delayed
platelet recovery. The use of PBSCs for allogeneic transplantation is also
being investigated. In allogeneic transplantation, donors are treated with
G-CSF in order to mobilize their PBSCs prior to
leukapheresis, the procedure that separates the fraction
containing stem cells from the other components in blood.
G-CSF is the cytokine most commonly
used for PBSC mobilization because of its increased efficacy and reduced toxicity
compared with GM-CSF. To mobilize stem cells, patients or donors are given 5–10
g/kg/d subcutaneously for 4 days. On the fifth day, they undergo leukapheresis.
The success of PBSC transplantation
depends upon transfusion of adequate numbers of stem cells. CD34, an antigen
present on early progenitor cells and absent from later, committed, cells, is
used as a marker for the requisite stem cells. The goal is to reinfuse at least
5 x 106 CD34 cells/kg; this number of CD34 cells usually results in prompt and
durable engraftment of all cell lineages. It can take several separate
leukaphereses to collect enough CD34 cells, especially from older patients and
patients who have been exposed to radiotherapy or chemotherapy.
Toxicity
Although the two growth factors have similar effects on
neutrophil counts, G-CSF is used more frequently because it is better
tolerated. G-CSF can cause bone pain, which clears when the drug is
discontinued. GM-CSF can cause more severe side effects, particularly at higher
doses. These include fevers, malaise, arthralgias, myalgias, and a capillary
leak syndrome characterized by peripheral edema and pleural or pericardial
effusions. Allergic reactions may occur but are infrequent. Splenic rupture is
a rare but serious complication of the use of G-CSF for PBSC.
Megakaryocyte Growth Factors
Chemistry
& Pharmacokinetics
Interleukin-11 (IL-11) is a 65–85
kDa protein produced by fibroblasts and stromal cells in the bone marrow. Oprelvekin,
the recombinant form of interleukin-11 approved for clinical use, is
produced by expression in E coli. The half-life of IL-11 is 7–8 hours
when the drug is injected subcutaneously.
Thrombopoietin, a 65–85
kDa glycosylated protein, is constitutively expressed by a variety of organs
and cell types. Hepatocytes appear to be the major source of human
thrombopoietin, and patients with cirrhosis and thrombocytopenia have low serum
thrombopoietin levels. Recombinant thrombopoietin is produced by expression in
human cells; the recombinant product contains two intramolecular disulfide
bonds and a number of carbohydrate side chains.
Pharmacodynamics
Interleukin-11
acts through a specific cell surface cytokine receptor to stimulate the growth
of multiple lymphoid and myeloid cells. It acts synergistically with other growth
factors to stimulate the growth of primitive megakaryocytic progenitors and,
most importantly, increases the number of peripheral platelets and neutrophils.
Acting
through its own cytokine receptor, thrombopoietin also independently stimulates
the growth of primitive megakaryocytic progenitors. In addition, it stimulates
mature megakaryocytes and even activates mature platelets to respond to
aggregation-inducing stimuli. The critical in vivo role of thrombopoietin has
been demonstrated in genetically engineered knockout mice who lack either
thrombopoietin or its receptor. These mice have marked thrombocytopenia but do
not display anemia or leukopenia.
Clinical
Pharmacology
Patients with thrombocytopenia have a high risk of
hemorrhage. While platelet transfusion is commonly used to treat
thrombocytopenia, this procedure can cause adverse reactions in the recipient;
furthermore, a significant number of patients fail to exhibit the expected
increase in platelet count.
Interleukin-11 is the first growth
factor to gain FDA approval for treatment of thrombocytopenia. It is approved
for the secondary prevention of thrombocytopenia in patients receiving
cytotoxic chemotherapy for treatment of nonmyeloid cancers. Clinical trials
show that it reduces the number of platelet transfusions required by patients
who experienced severe thrombocytopenia after a previous cycle of chemotherapy.
Although IL-11 has broad stimulatory effects on hematopoietic cell lineages in
vitro, it does not appear to have significant effects on the leukopenia or
neutropenia caused by myelosuppressive chemotherapy. Interleukin-11 is given by
subcutaneous injection at a
dose of 50 g/kg/d. It is started 6–24 hours after completion
of chemotherapy and continued for 14–21 days or until the platelet count passes
the nadir and rises to > 50,000 cells/ L.
Recombinant thrombopoietin is still
an investigational agent. The primary focus of current clinical trials is for
the treatment of chemotherapy-induced thrombocytopenia and thrombocytopenia
accompanying hematologic stem cell transplantation. Other trials are looking
into the possibility of administering thrombopoietin to normal donors in order
to increase the number of cells recovered by platelet apheresis. Approval of
the latter application will require that thrombopoietin be shown to have an
excellent short- and long-term safety profile.
Toxicity
The most common side effects of
interleukin-11 are fatigue, headache, dizziness, and
cardiovascular effects. The cardiovascular effects include anemia
(due to hemodilution), dyspnea (due to fluid accumulation in the lungs), and
transient atrial arrhythmias. Hypokalemia has also been seen in some patients.
All of these adverse effects appear to be reversible. In the limited clinical
trial data available thus far, recombinant thrombopoietin appears to be well
tolerated.

Drugs Used in Disorsers of Coagulation
Excessive bleeding and thrombosis
may represent altered states of hemostasis. Impaired hemostasis results in
spontaneous bleeding; stimulated hemostasis results in thrombus formation. The
drugs used to arrest abnormal bleeding and to inhibit thrombosis are the
subjects of this chapter.
Mechanisms
of Blood Coagulation
Thrombogenesis
Hemostasis is the spontaneous arrest
of bleeding from a damaged blood vessel. The normal vascular endothelial cell
is not thrombogenic, and circulating blood platelets and clotting factors do
not normally adhere to it to an appreciable extent.
The immediate hemostatic response of
a damaged vessel is vasospasm. Within seconds, platelets stick to the
exposed collagen of the damaged endothelium (platelet adhesion) and to
each other (platelet aggregation). Platelets then lose their individual
membranes and form a gelatinous mass during viscous metamorphosis. This platelet
plug quickly arrests bleeding but must be reinforced by fibrin for
long-term effectiveness. Fibrin reinforcement results from local stimuli to
blood coagulation: the exposed collagen of damaged vessels and the membranes
and released contents of platelets. The local production of thrombin not only
releases platelet adenosine diphosphate (ADP), a powerful inducer of
platelet aggregation, but also stimulates the synthesis of prostaglandins from
the arachidonic acid of platelet membranes. These powerful substances are
composed of two groups of eicosanoids that have opposite effects on
thrombogenesis.
Thromboxane A2 (TXA2)
is synthesized within platelets and induces thrombogenesis and
vasoconstriction. Prostacyclin (PGI2) is synthesized within vessel
walls and inhibits thrombogenesis. Serotonin (5-HT) is
also released from the platelets, stimulating further aggregation and
vasoconstriction.
The platelet is central to normal
hemostasis and to all thromboembolic disease. A white thrombus forms initially
in high-pressure arteries by adherence of circulating platelets to areas of
abnormal endothelium as described above. The growing thrombus of aggregated
platelets reduces arterial flow. This localized stasis triggers fibrin
formation, and a red thrombus forms around the nidal white thrombus.
Thrombogenesis
A red thrombus can form
around a white thrombus as mentioned above or de novo in low-pressure veins,
initially by adherence of platelets (as in arteries) but followed promptly by
the process of blood coagulation so that the bulk of the thrombus forms a long
tail consisting of a fibrin network in which red cells are enmeshed. These
tails become detached easily and travel as emboli to the pulmonary arteries.
Such emboli often arise from a deep venous thrombosis (DVT)—a thrombus in the
veins of the legs or pelvis. Although all thrombi are mixed, the platelet nidus
dominates the arterial thrombus and the fibrin tail the venous thrombus.
Arterial thrombi cause serious disease by producing local occlusive ischemia;
venous thrombi, by giving rise to distant embolization.
Blood
Coagulation
Blood coagulates by the
transformation of soluble fibrinogen into insoluble fibrin. Several circulating
proteins interact in a cascading series of limited proteolytic reactions. At
each step, a clotting factor zymogen (eg, factor VII) undergoes limited
proteolysis and becomes an active protease (eg, factor VIIa). Thus, each
protease factor activates the next clotting factor until finally a solid fibrin
clot is formed. Fibrinogen (factor I), the soluble precursor of fibrin, is the
substrate for the enzyme thrombin (factor IIa). This protease is formed during
coagulation by activation of its zymogen, prothrombin (factor II). Prothrombin
is bound by calcium to a platelet phospholipid (PL) surface, where activated
factor X (Xa), in the presence of factor Va, converts it into circulating
thrombin. Several of the blood clotting factors are targets for drug therapy.
The main initiator of blood coagulation
is the tissue factor (TF)/factor VIIa pathway. The exposure of TF on damaged
endothelium binds and activates circulating factor VII. This complex, in turn,
activates factors X and IX, with the eventual generation of thrombin. Thrombin,
in turn, activates upstream proteins, primarily factors V, VIII, and XI,
resulting in further thrombin generation. Additionally, thrombin is a potent
activator of platelets, converts fibrinogen to fibrin, and activates factor
XIII, resulting in an insoluble, cross-linked fibrin molecule.
The TF/factor VII/factor X process
is inhibited and regulated by tissue factor pathway inhibitor (TFPI). Oral
anticoagulant drugs inhibit the hepatic synthesis of several clotting factors.
Heparin inhibits the activity of several of these activated clotting factors by
enhancing the anticoagulant activity of antithrombin, which inactivates
the serine proteases IIa, IXa, Xa, XIa, and XIIa. The endogenous anticoagulants
protein C and protein S diminish amplification in the blood clotting
cascade by proteolysis of factors Va and VIIIa.
Regulation
of Coagulation & Fibrinolysis
Blood coagulation and thrombus
formation must be confined to the smallest possible area to achieve local
hemostasis in response to bleeding from trauma or surgery without causing
disseminated coagulation or impaired blood flow.

Two major
systems regulate and delineate these processes: fibrin inhibition and fibrinolysis.
Plasma
contains protease inhibitors that rapidly inactivate the coagulation proteins
as they escape from the site of vessel injury. The most important proteins of
this system are 1-antiprotease, 2- macroglobulin, 2-antiplasmin, and
antithrombin. If this system is overwhelmed, generalized intravascular clotting
may occur. This process is called disseminated intravascular coagulation
(DIC) and may follow massive tissue injury, cell lysis in malignant
neoplastic disease, obstetric emergencies such as abruptio placentae, or
bacterial sepsis.
The
central process of fibrinolysis is conversion of inactive plasminogen to the
proteolytic enzyme plasmin. Injured cells release activators of
plasminogen. Plasmin remodels the thrombus and limits the extension of
thrombosis by proteolytic digestion of fibrin.
Regulation
of the fibrinolytic system is useful in therapeutics. Increased fibrinolysis is
effective therapy for thrombotic disease. Tissue plasminogen activator
(t-PA), urokinase, and streptokinase all activate the fibrinolytic
system. Conversely, decreased fibrinolysis protects clots from lysis and
reduces the bleeding of hemostatic failure. Aminocaproic acid is a
clinically useful inhibitor of fibrinolysis. Heparin and the oral anticoagulant
drugs do not affect the fibrin-olytic mechanism.

Basic Pharmacology of the Anticoagulant Drugs
Indirect Thrombin Inhibitors
The
indirect thrombin inhibitors are so named because their antithrombotic effect
is exerted by their interaction with antithrombin. Unfractionated heparin
(UFH), low-molecular-weight heparin (LMWH), and the synthetic
pentasaccharide fondaparinux bind to antithrombin and enhance its
inactivation of factor Xa. UFH and to a lesser extent LMWH also enhance
antithrombin's inactivation of thrombin (IIa).
Heparin
Chemistry
& Mechanism of Action
Heparin is a heterogeneous mixture of
sulfated mucopolysaccharides. It binds to endothelial cell surfaces and a
variety of plasma proteins. As noted above, its biologic activity is dependent
upon the plasma protease inhibitor antithrombin. Antithrombin inhibits
clotting factor proteases, especially thrombin (IIa), IXa, and Xa, by forming
equimolar stable complexes with them. In the absence of heparin, these
reactions are slow; in the presence of heparin, they are accelerated 1000-
fold. Only about a third of the molecules in commercial heparin preparations
have an accelerating effect because the remainder lack the unique
pentasaccharide sequence needed for high-affinity binding to antithrombin. The
active heparin molecules bind tightly to antithrombin and cause a
conformational change in this inhibitor. The conformational change of
antithrombin exposes its active site for more rapid interaction with the
proteases (the activated clotting factors).
Heparin catalyzes the antithrombin-protease reaction without
being consumed. Once the antithrombin-protease complex is formed, heparin is
released intact for renewed binding to more antithrombin.
The antithrombin binding region of
commercial unfractionated heparin consists of repeating sulfated disaccharide
units composed of D-glucosamine-L-iduronic acid and D-glucosamine-Dglucuronic
acid. High-molecular-weight (HMW) fractions of heparin with high affinity for
antithrombin markedly inhibit blood coagulation by inhibiting all three
factors, especially thrombin and factor Xa.
The antithrombin binding region of
commercial unfractionated heparin consists of repeating sulfated disaccharide
units composed of D-glucosamine-L-iduronic acid and D-glucosamine-Dglucuronic
acid. High-molecular-weight (HMW) fractions of heparin with high affinity for
antithrombin markedly inhibit blood
coagulation by inhibiting all three factors, especially
thrombin and factor Xa. Unfractionated heparin has a MW range of 5000–30,000.
In contrast, the shorter-chain low-molecular-weight (LMW) fractions of heparin
inhibit activated factor X but have less effect on thrombin (and on coagulation
in general) than the HMW species. Nevertheless, numerous studies have
demonstrated that LMW heparins such as enoxaparin,
dalteparin, and tinzaparin are effective in several thromboembolic
conditions. In fact, these LMW heparins—in comparison with UFH—have
equal efficacy, increased bioavailability from the subcutaneous site of
injection, and less frequent dosing requirements (once or twice daily is
sufficient).
Because commercial heparin consists
of a family of molecules of different molecular weights, the correlation
between the concentration of a given heparin preparation and its effect on
coagulation often is poor. Therefore, UFH is standardized by bioassay. Heparin
sodium USP must contain at least 120 USP units per milligram. Heparin is
generally used as the sodium salt, but calcium heparin is equally effective.
Lithium heparin is used in vitro as an anticoagulant for blood samples.
Commercial heparin is
Heparin: origin, structure, and mechanism of action
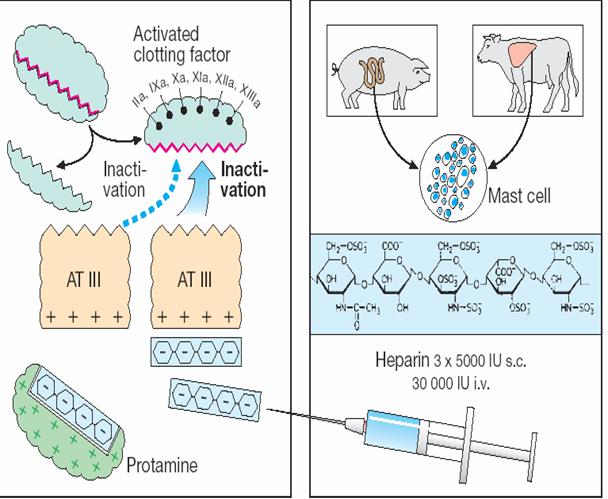
Heparin extracted from porcine
intestinal mucosa and bovine lung. Enoxaparin is obtained from the same sources
as regular heparin, but doses are specified in milligrams. Dalteparin,
tinzaparin and danaparoid (an LMW heparanoid containing heparan sulfate,
dermatan sulfate, and chondroitin sulfate that is no longer available in the
United States), on the other hand, are specified in anti-factor Xa units.
Toxicity
The major adverse effect of heparin
is bleeding. This risk can be decreased by scrupulous patient selection,
careful control of dosage, and close monitoring of the activated partial
thromboplastin time (aPTT) in those patients receiving unfractionated heparin.
Levels for UFH may also be determined by protamine titration (therapeutic
levels 0.2–0.4 unit/mL) or anti-Xa units (therapeutic levels 0.3–0.7 unit/mL).
Weight-based dosing of the LMW heparins results in predictable pharmacokinetics
and plasma levels in patients with normal renal function. Therefore, LMW
heparin levels are not generally measured except in the setting of renal
insufficiency, obesity, and pregnancy. LMW heparin levels are determined by
anti-Xa units. Peak therapeutic levels are 0.5–1 unit/mL for twice daily
dosing, determined 4 hours after administration, and approximately 1.5 units/mL
for once daily dosing. Elderly women and patients with renal failure are more
prone to hemorrhage. Heparin is of animal origin and should be used cautiously
in patients with allergy. Increased loss of hair and reversible alopecia have
been reported. Long-term heparin therapy is associated with osteoporosis and
spontaneous fractures. Heparin accelerates the clearing of postprandial lipemia
by causing the release of lipoprotein lipase from tissues, and long-term use is
associated with mineralocorticoid deficiency.

Heparin causes transient
thrombocytopenia in 25% or more of patients and severe thrombocytopenia in 5%. Mild
platelet reduction within the first 5 days of therapy may result from
heparin-induced aggregation that is postulated to be benign and transient in
character. A smaller subset of patients may develop an antibody-mediated
thrombocytopenia that is associated with paradoxical thrombosis. In these
instances, the heparin-induced antibody is directed against the heparin-platelet
factor 4 complex. These antigen-antibody complexes bind to Fc receptors on
adjacent platelets, causing aggregation and thromboembolism. The following
points should be considered in all patients receiving heparin: Platelet counts
should be performed frequently; thrombocytopenia should be considered to be
heparin-induced; any new thrombus can be the result of heparin; and
thromboembolic disease thought to be heparin-induced should be treated by
discontinuance of heparin and administration of an alternative drug, such as a
direct thrombin inhibitor (see below). Administration of warfarin alone is
contraindicated since it may exacerbatethe prothrombotic state associated with
heparin-induced thrombocytopenia.

Contraindications
Heparin is
contraindicated in patients who are hypersensitive to the drug, are actively
bleeding, or have hemophilia, significant thrombocytopenia, purpura, severe
hypertension, intracranial hemorrhage, infective endocarditis, active
tuberculosis, ulcerative lesions of the gastrointestinal tract, threatened
abortion, visceral carcinoma, or advanced hepatic or renal disease. Heparin
should be avoided in those patients who have recently had surgery of the brain,
spinal cord, or eye and in patients who are undergoing lumbar puncture or
regional anesthetic block. Despite the apparent lack of placental transfer,
heparin should be used in pregnant women only when clearly indicated.
Administration & Dosage
The
indications for the use of heparin are described in the section on clinical pharmacology.
A plasma concentration of heparin of 0.2–0.4 unit/mL (by protamine titration)
or 0.3–0.7 unit/mL (anti-Xa units) usually prevents pulmonary emboli in
patients with established venous thrombosis. This concentration of heparin will
prolong the activated partial thromboplastin time (aPTT) to 2–2.5 times that of
the control value. This degree of anticoagulant effect should be maintained
throughout the course of continuous intravenous heparin therapy. When intermittent
heparin administration is used, the aPTT should be measured 6 hours after
the administered dose to maintain prolongation of the aPTT to 2–2.5 times that
of the control value.
Hirudin
For a number of years, surgeons have
used medicinal leeches (Hirudo medicinalis) to prevent thrombosis in the
fine vessels of reattached digits. Hirudin is a specific, irreversible thrombin
inhibitor from the leech that is now available in recombinant form as lepirudin.
Its action is independent of antithrombin, which means it can reach and
inactivate fibrin-bound thrombin in thrombi. Lepirudin has little effect on
platelets or the bleeding time. Like heparin, it must be administered
parenterally and is monitored by the aPTT. Lepirudin is FDA-approved for use in
patients with thrombosis related to heparin-induced thrombocytopenia. This drug
has a short halflife, but it accumulates in renal insufficiency and no antidote
exists. Up to 40% of patients on longterm infusions develop an antibody
directed against the thrombin-lepirudin complex. These antigenantibody
complexes are not cleared by the kidney and may result in an enhanced
anticoagulant effect.
Bivalirudin, another
bivalent inhibitor of thrombin, is administered intravenously, with a rapid
onset and offset of action. The drug has a short half-life with clearance that
is 20% renal and the remainder metabolic. Bivalirudin inhibits platelet
activation and been FDA-approved for use in percutaneous coronary angioplasty.
Argatroban is a small
molecule thrombin inhibitor that is FDA approved for use in patients with
heparin-induced thrombocytopenia (HIT) with or without thrombosis and coronary
angioplasty in patients with HIT. It, too, has a short half-life, is given by
continuous intravenous infusion, and monitoring is done by aPTT. Its clearance
is not affected by renal disease but is dependent on liver function. The drug
requires dose reduction in patients with liver disease. Patients on argatroban
will demonstrate elevated INRs because of test interference, rendering the
transition to warfarin difficult.
Melagatran is the fourth parenteral
drug in this class. It and its oral prodrug, ximelagatran, are under intensive
study. Attractive features of ximelagatran include predictable pharmacokinetics
and bioavailability—allowing for fixed dosing and predictable anticoagulant
response; no need for routine coagulation monitoring; lack of interaction with
P450-interacting drugs; rapid onset and offset of action—allowing for immediate
anticoagulation and thus no need for overlap with additional anticoagulant
drugs. A published phase 3 trial in patients status post major orthopedic
surgery found ximelagatran equivalent to warfarin in preventing postoperative
DVT. Clinical trials in patients with acute DVT and chronic atrial fibrillation
are on-going.
Coumarin
Anticoagulants
Chemistry
& Pharmacokinetics
The clinical use of the coumarin
anticoagulants can be traced to the discovery of an anticoagulant substance
formed in spoiled sweet clover silage. It produced a deficiency of plasma
prothrombin and consequent hemorrhagic disease in cattle. The toxic agent was
identified as bishydroxycoumarin and synthesized as dicumarol. This drug and
its congeners, most notably warfarin, are widely used as rodenticides in
addition to their application as antithrombotic agents in humans. Warfarin is
the most reliable member of this group, and the other coumarin anticoagulants
are almost never used in the USA.
Warfarin is generally administered
as the sodium salt and has 100% bioavailability. Over 99% of racemic warfarin
is bound to plasma albumin, which may contribute to its small volume of
distribution (the albumin space), its long half-life in plasma (36 hours), and
the lack of urinary excretion of unchanged drug. Warfarin used clinically is a
racemic mixture composed of equal amounts of two enantiomorphs. The
levorotatory S-warfarin is four times more potent than the
dextrorotatory R-warfarin. This observation is useful in understanding
the stereoselective nature of several drug interactions involving warfarin.

Mechanism
of Action
Coumarin anticoagulants block the
-carboxylation of several glutamate residues in prothrombinand factors VII, IX,
and X as well as the endogenous anticoagulant proteins C and S.
The blockade results in incomplete molecules
that are biologically inactive in coagulation. This protein carboxylation is
physiologically coupled with the oxidative deactivation of vitamin K. The
anticoagulant prevents reductive metabolism of the inactive vitamin K epoxide
back to its active hydroquinone form. Mutational change of the responsible
enzyme, vitamin K epoxide reductase, can give rise to genetic resistance to
warfarin in humans and especially in rats.
There is an 8- to 12-hour delay in
the action of warfarin. Its anticoagulant effect results from a balance between
partially inhibited synthesis and unaltered degradation of the four vitamin
Kdependent clotting factors. The resulting inhibition of coagulation is
dependent on their degradation rate in the circulation. These half-lives are 6,
24, 40, and 60 hours for factors VII, IX, X, and II, respectively. Larger
initial doses of warfarin—up to about 0.75 mg/kg—hasten the onset of the
anticoagulant effect. Beyond this dosage, the speed of onset is independent of
the dose size. The only effect of a larger loading dose is to prolong the time
that the plasma concentration of drug remains above that required for
suppression of clotting factor synthesis. The only difference among oral
anticoagulants in producing and maintaining hypoprothrombinemia is the
half-life of each drug.
Toxicity
Warfarin
crosses the placenta readily and can cause a hemorrhagic disorder in the fetus.
Furthermore, fetal proteins with -carboxyglutamate residues found in bone and
blood may be affected by warfarin; the drug can cause a serious birth defect
characterized by abnormal bone formation. Thus, warfarin should never be
administered during pregnancy. Cutaneous necrosis with reduced activity of
protein C sometimes occurs during the first weeks of therapy. Rarely, the same
process causes frank infarction of breast, fatty tissues, intestine, and
extremities. The pathologic lesion associated with the hemorrhagic infarction
is venous thrombosis, suggesting that it is caused by warfarin-induced
depression of protein C synthesis.
The most serious interactions with
warfarin are those that increase the anticoagulant effect and the risk of
bleeding. The most dangerous of these interactions are the pharmacokinetic
interactions with the pyrazolones phenylbutazone and sulfinpyrazone. These
drugs not only augment the hypoprothrombinemia but also inhibit platelet
function and may induce peptic ulcer disease (see Chapter 36: Nonsteroidal
Anti-Inflammatory Drugs, Disease-Modifying Antirheumatic Drugs, Nonopioid
Analgesics, & Drugs Used in Gout).
The
mechanisms for their hypoprothrombinemic interaction are a stereoselective
inhibition of oxidative metabolic transformation of S-warfarin (the more
potent isomer) and displacement of albumin-bound warfarin, increasing the free
fraction. For this and other reasons, neither phenylbutazone nor sulfinpyrazone
is in common use in the USA. Metronidazole, fluconazole, and
trimethoprim-sulfamethoxazole also stereoselectively inhibit the metabolic
transformation of S-warfarin, whereas amiodarone, disulfiram, and
cimetidine inhibit metabolism of both enantiomorphs of warfarin. Aspirin,
hepatic disease, and hyperthyroidism augment warfarin
pharmacodynamically—aspirin by its effect on platelet function and the latter
two by increasing the turnover rate of clotting factors. The third-generation
cephalosporins eliminate the bacteria in the intestinal tract that produce
vitamin K and, like warfarin, also directly inhibit vitamin K epoxide
reductase. Heparin directly prolongs the prothrombin time by inhibiting the
activity of several clotting factors.
Barbiturates and rifampin cause a
marked decrease of the anticoagulant effect by induction of the hepatic
enzymes that transform racemic warfarin. Cholestyramine binds warfarin in the
intestine and reduces its absorption and bioavailability.
Pharmacodynamic reductions of
anticoagulant effect occur with vitamin K (increased synthesis of clotting
factors), the diuretics chlorthalidone and spironolactone (clotting factor
concentration), hereditary resistance (mutation of vitamin K reactivation cycle
molecules), and hypothyroidism (decreased turnover rate of clotting factors).
Drugs with no significant
effect on anticoagulant therapy include ethanol, phenothiazines,
benzodiazepines, acetaminophen, opioids, indomethacin, and most antibiotics.
Reversal
of Action
Excessive anticoagulant effect and
bleeding from warfarin can be reversed by stopping the drug and administering
vitamin K1 (phytonadione), fresh-frozen plasma, prothrombin complex
concentrates (PCC) such as Bebulin and Proplex T, and recombinant factor VIIa
(rFVIIa). The disappearance of excessive effect is not correlated with plasma
warfarin concentrations but rather with reestablishment of normal activity of
the clotting factors. A modest excess of anticoagulant effect without bleeding
may require no more than cessation of the drug. The warfarin effect can be
rapidly reversed in the setting of severe bleeding with the administration of a
prothrombin complex or recombinant factor VIIa coupled with intravenous vitamin
K.
Analogs
& Variants
Vitamin K antagonists other than warfarin are seldom used,
because they have less favorable pharmacologic properties or greater toxicity. Dicumarol
is incompletely absorbed and frequently causes gastrointestinal symptoms. Phenprocoumon
has a long half-life of 6 days—a disadvantage should toxicity occur. The
indanedione group, which includes phenindione and diphenadione, has potentially
serious adverse effects in the kidney and liver and is of little clinical use.
Basic
Pharmacology of the Fibrinolytic Drugs
Fibrinolytic
drugs rapidly lyse thrombi by catalyzing the formation of the serine protease plasmin
from its precursor zymogen, plasminogen. These drugs (streptokinase,
alteplase, anistreplase, tissue plasminogen activator, reteplase, tenecteplase,
and urokinase) create a generalized lytic state when administered
intravenously. Thus, both protective hemostatic thrombi and target
thromboemboli are broken down. The section Thrombolytic Drugs for Acute
Myocardial Infarction describes the use of these drugs in one major
application.
Pharmacology
Streptokinase is a protein (but not
an enzyme in itself) synthesized by streptococci that combines with the proactivator
plasminogen. This enzymatic complex catalyzes the conversion of inactive
plasminogen to active plasmin. Urokinase is a human enzyme synthesized
by the kidney that directly converts plasminogen to active plasmin. Plasmin
itself cannot be used because naturally occurring inhibitors in plasma prevent
its effects. However, the absence of inhibitors for urokinase and the
streptokinase-proactivator complex permit their use clinically. Plasmin formed
inside a thrombus by these activators is protected from plasma antiplasmins,
which allows it to lyse the thrombus from within.
Anistreplase (anisoylated
plasminogen streptokinase activator complex; APSAC) consists of a complex of
purified human plasminogen and bacterial streptokinase that has been acylated to
protect the enzyme's active site. When administered, the acyl group
spontaneously hydrolyzes, freeing the activated streptokinase-proactivator
complex. This product (recently discontinued in the USA) allows for rapid
intravenous injection, greater clot selectivity (ie, more activity on
plasminogen associated with clots than on free plasminogen in the blood), and
more thrombolytic activity.
Plasminogen can also be activated
endogenously by tissue plasminogen activators (t-PA). These activators
preferentially activate plasminogen that is bound to fibrin, which (in theory)
confines fibrinolysis to the formed thrombus and avoids systemic activation.
Human t-PA is manufactured as alteplase by means of recombinant DNA technology.
Reteplase is another
recombinant human t-PA from which several amino acid sequences have been
deleted. Reteplase is less expensive to produce than t-PA. Because it lacks the
major fibrin-binding domain, reteplase is less fibrin-specific than t-PA. Tenecteplase
is a mutant form of t-PA that has a longer half-life, and it can be given
as an intravenous bolus. Tenecteplase is slightly more fibrinspecific than
t-PA.
Indications
& Dosage
Use of fibrinolytic drugs by the intravenous route is
indicated in cases of multiple pulmonary emboli that are not massive
enough to require surgical management. Intravenous fibrinolytic drugs are also
indicated in cases of central deep venous thrombosis such as the
superior vena caval syndrome and ascending thrombophlebitis of the iliofemoral
vein. They have also been used intraarterially, especially for peripheral
vascular disease.
Thrombolytic
therapy in the management of acute myocardial infarction requires
careful patient selection, the use of a specific thrombolytic agent, and the
benefit of adjuvant therapy. Considerable controversy surrounds the question of
greater safety or efficacy of t-PA compared with the other thrombolytic agents
.
Streptokinase
is administered by intravenous infusion of a loading dose of 250,000 units,
followed by 100,000 units/h for 24–72 hours.
Patients with antistreptococcal
antibodies can develop fever, allergic reactions, and therapeutic resistance.
Urokinase requires a loading dose of 300,000 units given over 10 minutes and a
maintenance dose of 300,000 units/h for 12 hours. Alteplase (t-PA) is given by
intravenous infusion of 60 mg over the first hour and then 40 mg at a rate of
20 mg/h. Reteplase is given as two intravenous bolus injections of 10 units
each, separated by 30 minutes. Tenecteplase is given as a single intravenous
bolus of 0.5 mg/kg. Anistreplase is given as a single intravenous injection of
30 units over 3–5 minutes. A single course of fibrinolytic drugs is expensive:
hundreds of dollars for streptokinase and thousands for urokinase and t-PA.
Recombinant tissue plasminogen
activator has increasingly been used for patients presenting with acute stroke
symptoms. A recent outcomes study demonstrated an advantage with respect to
neurologic disability at 1 year in those patients with acute ischemic stroke
who received intravenous t-PA within 3 hours after onset of symptoms.

Thrombolytic
Drugs for Acute Myocardial Infarction
Background: The
paradigm shift in 1980 on the causation of acute myocardial infarction to acute
coronary occlusion by a thrombus created the rationale for thrombolytic therapy
of this common lethal disease. At that time—and for the first time—intravenous
thrombolytic therapy for acute myocardial infarction in the European
Cooperative Study Group trial was found to reduce mortality significantly.
Later studies, with thousands of patients in each trial, provided enough
statistical power for the 20% reduction in mortality to be considered
statistically significant. Although the standard of care in areas with adequate
facilities and experience in percutaneous coronary intervention (PCI) now
favors catheterization and placement of a stent, thrombolytic therapy is still
very important where PCI is not readily available.
Patients: The selection
of patients for thrombolytic therapy is critical. The diagnosis of acute
myocardial infarction is made clinically and is confirmed by
electrocardiography. Patients with ST segment elevation and bundle branch block
on electrocardiography do best; those with ST segment depression or a normal
ECG do less well; and those with non-Q-wave acute myocardial infarction may
even be harmed. All trials to date show the greatest benefit for thrombolytic
therapy when it is given early, within 6 hours after symptomatic onset of acute
myocardial infarction.
Clinical trials: One of the
trials (ISIS-3) showed that streptokinase plus aspirin performed as well as
recombinant tissue-type plasminogen activator (rt-PA) or complex formulations
of streptokinase such as anistreplase (APSAC). The GUSTO trial showed a small
advantage for the much more expensive t-PA over streptokinase, but with a
significantly higher risk of hemorrhagic stroke. Nine clinical trials—each
containing over 1000 patients with suspected acute myocardial infarction—
reported 11.6% mortality at 35 days in the control group and 9.5% in the
treatment group, an 18% reduction in mortality.
Adjunctive drugs: The best
results occur with thrombolytic drugs supplemented by other drugs. - Blocker
drugs reduced myocardial ischemia and infarct size, prevented arrhythmias,
decreased reinfarction, and improved survival in ISIS-1 and GISSI-1. Aspirin
alone and with streptokinase reduced mortality in ISIS-2. Nitroglycerin, given
early in acute myocardial infarction for pain relief, had no beneficial effect
on mortality in GISSI-3 and ISIS-4. Oral anticoagulants are best used in
patients with depressed left ventricular function or systemic embolization.
Heparin was a necessary adjunct for t-PA in GUSTO but was less useful with
streptokinase plus aspirin because of the increased risk of bleeding. ACE
inhibitors reduced infarct expansion and arrhythmias and improved survival in
GISSI-3. Direct thrombin inhibitors like hirudin and bivalirudin are undergoing
clinical trials to better determine their efficacy in conjunction with
thrombolytic therapy (TIMI-9B, GUSTO-2A, OASIS, HERO).
Summary: Thrombolytic
drugs reduce the mortality of acute myocardial infarction. The early and
appropriate use of any thrombolytic drug probably transcends individual
advantages. Adjunctive drugs like aspirin, -blockers, and ACE inhibitors reduce
mortality even further. The principles of management are outlined in part 7 of
the American Heart Association Guidelines, 2000.
Basic
Pharmacology of Antiplatelet Agents
Platelet
function is regulated by three categories of substances. The first group
consists of agents generated outside the platelet that interact with platelet
membrane receptors, eg, catecholamines, collagen, thrombin, and prostacyclin. The
second category contains agents generated within the platelet that interact
with membrane receptors, eg, ADP, prostaglandin D2, prostaglandin E2, and
serotonin. The third group comprises agents generated within the platelet that
act within the platelet, eg, prostaglandin endoperoxides and thromboxane A2,
the cyclic nucleotides cAMP and cGMP, and calcium ion. From this list of
agents, several targets for platelet inhibitory drugs have been identified:
inhibition of prostaglandin metabolism (aspirin), inhibition of ADPinduced
platelet aggregation (clopidogrel, ticlopidine), and blockade of GP IIb/IIIa
receptors on platelets (abciximab, tirofiban, and eptifibatide). Dipyridamole
and cilostazol are additional antiplatelet drugs.
Aspirin
The prostaglandin thromboxane A2
is an arachidonate product that causes platelets to change shape, to release
their granules, and to aggregate (see Chapter 18: The Eicosanoids:
Prostaglandins, Thromboxanes, Leukotrienes, & Related Compounds). Drugs
that antagonize this pathway interfere with platelet aggregation in vitro and
prolong the bleeding time in vivo. Aspirin is the prototype of this
class of drugs. Drugs that modulate the intraplatelet concentration of cAMP do
not prolong the bleeding time. However, dietary therapy can be useful in the
prophylaxis of thrombosis. Ingestion of the unsaturated fatty acid eicosapentaenoic
acid, which is high in cold water fish, generates prostaglandin I3, an
effective antiaggregating substance like prostacyclin, and thromboxane A3,
which is much less active than TXA2.
Related Compounds, aspirin inhibits
the synthesis of thromboxane A2 by irreversible acetylation of the enzyme
cyclooxygenase. Other salicylates and nonsteroidal anti-inflammatory drugs also
inhibit cyclooxygenase but have a shorter duration of inhibitory action because
they cannot acetylate cyclooxygenase, ie, their action is reversible.
The FDA has approved the use of 325
mg/d for primary prophylaxis of myocardial infarction but urges caution
in this use of aspirin by the general population except when prescribed as an
adjunct to risk factor management by smoking cessation and lowering of blood
cholesterol and blood pressure. Meta-analysis of many published trials of
aspirin and other antiplatelet agents confirms the value of this intervention
in the secondary prevention of
vascular events among patients with a history of vascular events.

Inhibitors of platelet aggregation
Clopidogrel
& Ticlopidine
Clopidogrel and ticlopidine reduce platelet aggregation by
inhibiting the ADP pathway of platelets. These drugs are thienopyridine
derivatives that achieve their antiplatelet effects by irreversibly blocking
the ADP receptor on platelets. Unlike aspirin, these drugs have no effect on
prostaglandin metabolism. Randomized clinical trials with both drugs report
efficacy in the prevention of vascular events among patients with transient
ischemic attacks, completed strokes, and unstable angina pectoris. Use of
clopidogrel or ticlopidine to prevent thrombosis is now considered standard practice
in patients undergoing placement of a coronary stent.
Presystemic inactivation of
platelet cyclooxygenase by acetylsalicylic acid
Adverse effects of ticlopidine
include nausea, dyspepsia, and diarrhea in up to 20% of patients, hemorrhage in
5%, and, most seriously, leukopenia in 1%. The leukopenia is detected by
regular monitoring of the white blood cell count during the first 3 months of
treatment. Development of thrombotic thrombocytopenic purpura (TTP) has also
been associated with the ingestion of ticlopidine. The dosage of ticlopidine is
250 mg twice daily. It is particularly useful in patients who cannot tolerate
aspirin. Doses of ticlopidine less than 500 mg/d may be efficacious with fewer
adverse effects.
Clopidogrel has fewer adverse effects
than ticlopidine and is rarely associated with neutropenia. Thrombotic
thrombocytopenic purpura associated with clopidogrel has recently been
reported. Because of its superior side effect profile and dosing requirements,
clopidogrel is preferred over ticlopidine. The antithrombotic effects of
clopidogrel are dose-dependent; within 5 hours after an oral loading dose of
300 mg, 80% of platelet activity will be inhibited. The maintenance dose of
clopidogrel is 75 mg/d, which achieves maximum platelet inhibition. The
duration of the antiplatelet effect is 7–10 days.
Blockade
of Platelet Gp IIb/IIIa Receptors
The glycoprotein IIb/IIIa inhibitors are used in patients
with acute coronary syndromes. These drugs target the platelet IIb/IIIa receptor
complex. The IIb/IIIa complex functions as a receptor mainly for fibrinogen and
vitronectin but also for fibronectin and von Willebrand factor. Activation of
this receptor complex is the "final common pathway" for platelet
aggregation. There are approximately 50,000 copies of this complex on the
platelet surface. Persons lacking this receptor have a bleeding disorder called
Glanzmann's thrombasthenia.
Abciximab, a humanized monoclonal antibody
directed against the IIb/IIIa complex including the vibronectin receptor, was
the first agent approved in this class of drugs. It has been approved for use
in percutaneous coronary intervention and in acute coronary syndromes. Eptifibatide
is an analog of the sequence at the extreme carboxyl terminal of the delta
chain of fi-brinogen, which mediates the binding of fibrinogen to the receptor.
Tirofiban is a smaller molecule with similar properties. Eptifibatide
and tirofiban inhibit ligand binding to the IIb/IIIa receptor by their
occupancy of the receptor but do not block the vibronectin receptor. The three
agents described above are administered parenterally. Oral formulations of
IIb/IIIa antagonists have been developed and are in various stages of
development. Thus far, however, lack of efficacy and significant
thrombocytopenia have prevented progress with the oral analogs.
Additional
Antiplatelet-Directed Drugs
Dipyridamole is a
vasodilator that inhibits platelet function by inhibiting adenosine uptake and
cyclic GMP phosphodiesterase activity. Dipyridamole by itself has little or no
beneficial effect. Therefore, therapeutic use of this agent is primarily in
combination with aspirin to prevent cerebrovascular ischemia. It may also be
used in combination with warfarin for primary prophylaxis of thromboemboli in
patients with prosthetic heart valves. A combination of dipyridamole complexed
with 25 mg of aspirin is now available for secondary prophylaxis of
cerebrovascular disease.
Cilostazol is a newer
phosphodiesterase inhibitor that promotes vasodilation and inhibition of
platelet aggregation. Cilostazol is used primarily to treat intermittent
claudication.
Clinical
Pharmacology of Drugs Used to Prevent Clotting
Venous
Thrombosis
Risk Factors
Inherited Disorders
The inherited disorders
characterized by an tendency to form thrombi (thrombophilia) derive from either
quantitative or qualitative abnormalities of the natural anticoagulant system.
Deficiencies in the natural anticoagulants antithrombin, protein C, and protein
S account for approximately 15% of selected patients with juvenile or recurrent
thrombosis and 5–10% of unselected cases of acute venous thrombosis. Additional
causes of thrombophilia include the factor V Leiden mutation,
hyperhomocystinemia, and the prothrombin 20210 mutation that together account
for the greater number of hypercoagulable patients.
Acquired
Disease
The increased risk of thromboembolism associated with
arrhythmia, primarily atrial fibrillation, and the placement of mechanical heart
valves has long been recognized. Similarly, prolonged bed rest, high-risk
surgical procedures, and the presence of cancer are clearly associated with an
increased incidence of deep venous thrombosis and embolism.
Antithrombotic Management
Prevention
Primary prevention of venous
thrombosis reduces the incidence of and mortality rate from pulmonary emboli.
Heparin and warfarin may be used to prevent venous thrombosis. Subcutaneous
administration of low-dose unfractionated heparin, low-molecular-weight heparin,
or fondaparinux provides effective prophylaxis. Warfarin is also effective but
requires laboratory monitoring of the prothrombin time.
Treatment
of Established Disease
Treatment for established venous
thrombosis is initiated with unfractionated or low-molecularweight heparin for
the first 5–7 days, with an overlap with warfarin. Once therapeutic effects of
warfarin have been established, therapy with warfarin is continued for a
minimum of 3–6 months. Patients with recurrent disease or identifiable, nonreversible
risk factors may be treated indefinitely. Small thrombi confined to the calf
veins may be managed without anticoagulants if there is documentation over time
that the thrombus is not extending. Warfarin readily crosses the placenta. It
can cause hemorrhage at any time during pregnancy as well as developmental
defects when administered during the first trimester. Therefore, venous
thromboembolic disease in pregnant women is generally treated with heparin,
best administered by subcutaneous injection.
Arterial Thrombosis
Activation of platelets is
considered an essential process for arterial thrombosis. Thus, treatment with
platelet-inhibiting drugs such as aspirin and ticlopidine or clopidogrel is
indicated in patients with transient ischemic attacks and strokes or unstable
angina and acute myocardial infarction. In angina and infarction, these drugs
are often used in conjunction with -blockers, calcium channel blockers, and
fibrinolytic drugs.
Drugs Used in Bleeding Disorders
VItamin
K
Vitamin K confers biologic activity upon prothrombin and
factors VII, IX, and X by participating in their postribosomal modification.
Vitamin K is a fat-soluble substance found primarily in leafy green vegetables.
The dietary requirement is low, because the vitamin is additionally synthesized
by bacteria that colonize the human intestine. Two natural forms exist:
vitamins K1 and K2. Vitamin K1 (phytonadione); is found in food. Vitamin K2
(menaquinone) is found in human tissues and is synthesized by intestinal bacteria. Vitamins K1 and K2 require bile salts for
absorption from the intestinal tract. Vitamin K1 is available clinically in 5
mg tablets and 50 mg ampules. Onset of effect is delayed for 6 hours but the
effect is complete by 24 hours when treating depression of prothrombin activity
by excess warfarin or vitamin K deficiency. Intravenous administration of
vitamin K1 should be slow, because rapid infusion can produce dyspnea, chest
and back pain, and even death. Vitamin K repletion is best achieved with intravenous
or oral administration, because its bioavailability after subcutaneous
administration is erratic.
Vitamin K1 is currently administered
to all newborns to prevent the hemorrhagic disease of vitamin K deficiency,
which is especially common in premature infants. The water-soluble salt of
vitamin K3 (menadione) should never be used in therapeutics. It is particularly
ineffective in the treatment of warfarin overdosage.
Vitamin K deficiency frequently
occurs in hospitalized patients in intensive care units because of poor diet,
parenteral nutrition, recent surgery, multiple antibiotic therapy, and uremia.
Severe hepatic failure results in diminished protein synthesis and a
hemorrhagic diathesis that is unresponsive to vitamin K.
Plasma Fractions
Sources
& Preparations
Deficiencies in plasma coagulation
factors can cause bleeding. Spontaneous bleeding occurs when factor activity is
less than 5–10% of normal. Factor VIII deficiency (classic hemophilia, or
hemophilia A) and factor IX deficiency (Christmas disease, or hemophilia B)
account for most of the heritable coagulation defects. Concentrated plasma
fractions are available for the treatment of these deficiencies. Administration
of plasma-derived, heat- or detergent-treated factor concentrates and recombinant
factor concentrates are the standard treatments for bleeding associated with
hemophilia. Lyophilized factor VIII concentrates are prepared from large pools
of plasma. Transmission of viral diseases such as hepatitis B and C and AIDS is
reduced or eliminated by pasteurization and by extraction of plasma with
solvents and detergents. The best use of these therapeutic materials requires
diagnostic specificity of the deficient factor and quantitation of its activity
in plasma. Intermediate purity factor VIII concentrates (as opposed to
recombinant or highpurity concentrates) contain significant amounts of von
Willebrand factor. Humate-P is a factor VIII concentrate that is approved by
the FDA for the treatment of bleeding associated with von Willebrand disease.
Vitamin
K-antagonists of the coumarin type and vitamin K
Clinical
Uses
An uncomplicated hemorrhage into a
joint should be treated with sufficient factor VIII or factor IX replacement to
maintain a level of at least 30–50% of the normal concentration for 24 hours.
Soft tissue hematomas require a minimum of 100% activity for 7 days. Hematuria
requires at least 10% activity for 3 days. Surgery and major trauma require a
minimum of 100% activity for 10 days. The initial loading dose for factor VIII
is 50 units/kg of body weight to achieve 100% activity of factor VIII from a
baseline of 1%, assuming a normal hemoglobin. Each unit of factor VIII per
kilogram of body weight raises its activity in plasma 2%. Replacement should be
administered every 12 hours. Factor IX therapy requires twice the dose of
factor VIII, but with an administration of about every 24 hours because of its
longer half-life. Recombinant factor IX has only 80% recovery compared to
plasma-derived factor IX products. Therefore, dosing with recombinant factor IX
requires 120% of the dose used with the plasma-derived product.
Desmopressin acetate (arginine
vasopressin) increases the factor VIII activity of patients with mild
hemophilia A or von Willebrand disease. It can be used in preparation for minor
surgery such as tooth extraction without any requirement for infusion of
clotting factors if the patient has a documented adequate response. High-dose
intranasal desmopressin is available and has been shown to be efficacious and
well tolerated by patients.
Freeze-dried concentrates of plasma
containing prothrombin, factors IX and X, and varied amounts of factor VII
(Proplex, etc) are commercially available for treating deficiencies of these
factors. Each unit of factor IX per kilogram of body weight raises its activity
in plasma 1.5%. Heparin is often added to inhibit coagulation factors activated
by the manufacturing process. However, addition of heparin does not eliminate
all thromboembolic events.
Some preparations of factor IX concentrate contain activated
clotting factors, which has led to their use in treating patients with
inhibitors or antibodies to factor VIII or factor IX. Two products are
available expressly for this purpose: Autoplex (with factor VIII
correctional activity) and Feiba (with factor VIII inhibitor bypassing
activity). These products are not uniformly successful in arresting hemorrhage,
and the factor IX inhibitor titers often rise after treatment with them.
Acquired inhibitors of coagulation factors may also be treated with porcine
factor VIII (for factor VIII inhibitors) and recombinant activated factor VII.
Recombinant activated factor VII (NovoSeven) is being increasingly used
to treat coagulopathy associated with liver disease and major blood loss in
trauma and surgery. These recombinant and plasma-derived factor concentrates
are very expensive, and the indications for them are very precise. Therefore,
close consultation with a hematologist knowledgeable in this area is essential.
Cryoprecipitate
is
a plasma protein fraction obtainable from whole blood. It is used to treat
deficiencies or qualitative abnormalities of fibrinogen, such as that which
occurs with disseminated intravascular coagulation and liver disease. A single
unit of cryoprecipitate contains 300 mg of fibrinogen.
Cryoprecipitate
may also be used for patients with factor VIII deficiency and von Willebrand
disease if desmopressin is not indicated and a pathogen-inactivated recombinant
or plasma-derived product is not available. The concentration of factor VIII
and von Willebrand factor in cryoprecipitate is not as great as that found in
the concentrated plasma fractions. Moreover, cryoprecipitate is not treated in
any manner to decrease the risk of viral exposure. For infusion, the frozen
cryoprecipitate unit is thawed and dissolved in a small volume of sterile
citrate-saline solution and pooled with other units. Rh-negative women with
potential for childbearing should receive only Rh-negative cryoprecipitate because
of possible contamination of the product with Rhpositive blood cells.
Fibrinolytic
Inhibitors: Aminocaproic Acid
Aminocaproic acid (EACA), which is
chemically similar to the amino acid lysine, is a synthetic inhibitor of
fibrinolysis. It competitively inhibits plasminogen activation. It is rapidly
absorbed orally and is cleared from the body by the kidney. The usual oral
dosage of EACA is 6 g four times a day. When the drug is administered
intravenously, a 5 g loading dose should be infused over 30 minutes to avoid
hypotension. Tranexamic acid is an analog of aminocaproic acid and has
the same properties. It is administered orally with a 15 mg/kg loading dose
followed by 30 mg/kg every 6 hours, but the drug is not currently available in
the United States.
Clinical uses of aminocaproic acid
are as adjunctive therapy in hemophilia, as therapy for bleeding from
fibrinolytic therapy, and as prophylaxis for rebleeding from intracranial
aneurysms. Treatment success has also been reported in patients with postsurgical
gastrointestinal bleeding and postprostatectomy bleeding and bladder hemorrhage
secondary to radiation- and drug-induced cystitis. Adverse effects of the drug
include intravascular thrombosis from inhibition of plasminogen activator,
hypotension, myopathy, abdominal discomfort, diarrhea, and nasal stuffiness.
The drug should not be used in patients with disseminated intravascular
coagulation or genitourinary bleeding of the upper tract, eg, kidney and
ureters, because of the potential for excessive clotting.
Serine Protease Inhibitors: Aprotinin
Aprotinin
is a serine protease inhibitor ("serpin") that inhibits fibrinolysis
by free plasmin and may have other antihemorrhagic effects as well. It also
inhibits the plasmin-streptokinase complex in patients who have received that
thrombolytic agent. Aprotinin will reduce bleeding—by as much as 50%—from many
types of surgery, especially that involving extracorporeal circulation for open
heart procedures and liver transplantation. It is currently approved for use in
patients undergoing coronary artery bypass grafting who are at high risk of
excessive blood loss. In placebo-controlled trials, adverse effects of
aprotinin were little different from those reported in patients in the placebo
group. In larger studies, a possible association with anaphylaxis has been
reported in < 0.5% of cases. Therefore, a small test dose is recommended
before the full therapeutic dose is given.
ANTICOAGULANT THERAPY
Thrombosis:
· The formation
of clots at the wrong time in the wrong place
· Can be
arterial or venous
Risk
factors are different:
VENOUS ARTERIAL
immobility smoking
obesity male
sex
trauma/surgery hypertension
cancer hyperlipidaemia
pregnancy diabetes
oral contraceptive raised
fibrinogen
thrombophilia inherited
factors
increasing age increasing
age
Complications
include:
Venous:
·
Deep
vein thrombosis (DVT)
· Pulmonary
embolism (PE)
· Postphlebitic
syndrome
Arterial:
· Myocardial
infarction (coronary thrombosis)
· Stroke
(cerebral thrombosis)
· Peripheral
arterial occlusion (infarction, gangrene)
Krysko
A.A., Malovichko O.L., Kabanova T.A. and Mazepa A.V. Russian Journal of
Bioorganic Chemistry, 2004, 30,
534.
ANTICOAGULANT THERAPY
Two
main indications:
· Prevention
of thrombosis (prophylaxis)
· Treatment
of thrombosis
3 main drugs are used:
· Aspirin
· Heparins
· Warfarin
1.
ASPIRIN
q Mainly used for the prevention of arterial thrombosis
q Is an “anti-platelet” drug, ie inhibits platelet function
q Works by irreversibly inactivating cyclo-oxygenase and reducing the formation of thromboxane (activates and aggregates platelets)
q Usual dose 75mg (“mini-dose aspirin”)
q Prevention can be:
· “PRIMARY”
in
patients known to be at high risk but have been OK so far
· or
“SECONDARY”
to prevent
further problems in a patient who has
already had a thrombotic event such as MI or stroke
2.
HEPARINS
q These are naturally occurring glycosaminoglycans produced by
cells such as vascular endothelium
q Work by binding to the natural anticoagulant “Antithrombin” and increasing its ability
to inhibit several points in the clotting cascade, especially Xa and
Thrombin
q Not active orally and has to be given by Intravenous or
Subcutaneous injection
q Main uses:
· Initial
treatment of acute venous or arterial thrombosis
· Prophylaxis
during major surgery
· Prophylaxis
in pregnancy (where warfarin is contraindicated)
q 2 main types of heparin:
· Unfractionated
heparin (UFH)
- usually
given by IV infusion
- narrow “therapeutic
window” between anticoagulant effect and risk of bleeding
- requires
laboratory monitoring with aim of
keeping APTT between 1.5 and 2.5 x normal
· Low
molecular weight heparins
- made by
enzymatic digestion of UFH
- main
therapeutic effect is against Factor Xa
- bigger
therapeutic window, lower risk of bleeding
- do not
usually need laboratory monitoring (prescribed on a dose per weight basis as
have a much more predictable effect than UFH)
- usually
given by subcutaneous injection and patients can be taught to self-inject
3.
WARFARIN
q Works by inhibiting the formation of those clotting factors
which require Vitamin K for full activation:
-
Factors
II, VII, IX and X
q Monitored by measuring the Prothrombin Time (standardised
throughout the world as the:
International
Normalised Ratio (INR))
q Main side-effect is bleeding; depends on degree of
anticoagulation, duration of treatment, quality of control (patients usually
attend a special Anticoagulant Service), drug interactions (common in elderly)
q Orally administered
q Takes several days to achieve a full anticoagulant effect
therefore, for treatment of thrombosis, usually start both heparin
(instantaneous effect) and warfarin together and stop the heparin when reach
therapeutic INR
q INDICATIONS RECOMMENDED INR
Treatment of DVT and/or PE 2.0-3.0
Atrial Fibrillation 2.0-3.0
Recurrent DVT and/or PE 3.0-4.5
Metal heart valves 3.0-4.5
q Duration of treatment:
· Limited
DVT with reversible 6 weeks
risk
factor (eg surgery)
· More
extensive DVT or 3 to 6
months
Pulmonary
embolism
· Long term
risk factors
eg Atrial
Fibrillation, metal Lifelong
heart valve,
recurrent thrombosis
Control
of warfarin treatment:
· Patients
usually attend an Anticoagulant Clinic
(now
usually “community based”, eg at Health Centres)
· Initially
need frequent checks on INR
· Once
stabilised can go for up to 12 weeks between checks
· Many
clinics now use computer-assisted dosing (more standardised than dosing by
doctors or nurses, but needs watching intelligently!)
· Some
patients buy home coagulation monitors and can download results to central
clinic for computer-dosing via e-mail
· Biggest
problems occur in the elderly, those on multiple drugs and the poorly compliant
patient
1.
http://www.youtube.com/watch?v=8W0UzjmSPWc&feature=related
2.
http://www.youtube.com/watch?v=gGrDAGN5pC0&feature=related
3.
http://www.youtube.com/watch?v=--bZUeb83uU&feature=related
4.
http://www.youtube.com/watch?v=WXOBJEXxNEo&feature=related
5. http://www.youtube.com/watch?v=Lt0BjNwF6IU&feature=related
6.
http://www.youtube.com/watch?v=fjjEiVeYRNg&feature=related
7.
http://www.youtube.com/watch?v=mut1VYu9tzk&feature=related
8.
http://www.youtube.com/watch?v=6zEiH7X1Jz4&NR=1
9.
http://www.youtube.com/watch?v=kUiEEski4Aw&feature=related