Manifestations in the oral cavity in diseases
of the blood in children. Tactics children's dentist.
Red and white
blood cells are the cornerstones of systemic health, and disorders of these
cells may have profound effects on all organ systems. In the periodontium, red
blood cells carry oxygen and nutrients to the tissues. Platelets are critical
for hemostasis and for production of critical immunoinflammatory mediators.
White blood cells protect the periodontium and oral mucosa from bacterial,
viral, and fungal pathogens. These cells exist in an exquisite homeostatic
balance. Disorders that disturb this balance may manifest within the
periodontium or on other mucosal surfaces.
Oral signs and symptoms may be the first clinical evidence of underlying
blood disorders, and the dental health practitioner must pay careful attention
to possible indicators of occult disease. Diseases of blood frequently affect soft and
hard tissues of mouth with different characteristics. Oral manifestation of
blood disease affect color of the mucosa hypertrophy of gingiva, mucosal
destruction in the form of ulceration, bleeding and hemorrhage, color of the
tooth with red discoloration, lymph node and affect the bone with decreased
density and enlarged marrow space. Every dentist should be familiarized the
wide variety of oral manifestations of the blood diseases to differentiate from
other diseases.
An abnormal blood count or blood cell
morphology does not necessarily indicate a primary haematology problem because
it may reflect an underlying non-haematological condition or may be the result
of therapeutic interventions. Anaemia occurs in many conditions, but a primary
blood disease should be considered when a patient has splenomegaly; lymphadenopathy;
a bleeding tendency or thrombosis; and/or non-specific symptoms (malaise,
sweats, or weight loss). As with any clinical problem, the first steps in determining
the diagnosis include obtaining a care-ful clinical and drug history and
thorough physical examination. The result of these, in combination with the patient’s age, sex, ethnic origin,
social and family history, and knowledge of the locally prevalent diseases,
will determine subsequent laboratory investigations.
The investigation of specific
haematological prob-lems is covered in detail in Chapters 7 (iron deficiency
anaemia), 8 (megaloblastic anaemia), 9, 10, and 11 (haemolytic anaemias), 12
(haemoglo-binopathies), and 16 and 17 (coagulation disorders).
Interpretation of
Screening Tests
Results of laboratory screening tests
should always be interpreted with an understanding of the limi-tations of the
tests and the physiological variations that occur with sex, age, and conditions
such as pregnancy and exercise. Physiological variations in cell counts are
detailed in Chapter 2. Abnormalities of red cells, white cells, or platelets
may be quanti-tative (increased or reduced numbers) or qualitative (abnormal
appearance and/or function).
Increases Affecting More Than One Cell
Line
A simultaneous increase in the cells of
more than one cell line suggests that the overproduction of cells originates in
an early precursor cell. This occurs in myeloproliferative disorders in which
one cell type may predominate, e.g., platelets in essential thrombocythaemia
and red cells in polycythaemia vera (primary proliferative polycythaemia), but
there are often increases in other cell lines. The diagnosis will depend on
which cell line expansion is dominant.
Erythrocytosis
Increases in red cells may be one of the
following:
• “Relative” (pseudopolycythaemia) owing
to reduced plasma volume
• “Primary” (polycythaemia vera) as part
of the spectrum of myeloproliferative disorders
• “Secondary” to chronic hypoxia (e.g.,
chronic lung disease, congenital heart disease, high-affinity hae-moglobins) or
aberrant erythropoietin production
Secondary polycythaemia can generally be excluded
by the clinical history and examination, assessment of serum erythropoietin
concentration and arterial oxygen saturation, haemoglobin electrophoresis, and
abdominal ultrasound. The presence of splenomegaly is suggestive of
poly-cythaemia vera, and this diagnosis can be confirmed by demonstrating an
absolute increase in total red cell volume and excluding other causes of
erythro-cytosis. Measurement of red cell and/or plasma volume will identify
pseudopolycythaemia.
Leucocytosis
Neutrophilia
Neutrophils are commonly increased in
number during pregnancy and in acute infections, inflam-mation, intoxication,
corticosteroid therapy, and acute blood loss or destruction. Neutrophilia with the
neutrophils showing heavy cytoplasmic granu-lation (“toxic” granulation) is a
common finding in severe bacterial infections. In the absence of any underlying
cause, a high neutrophil count with
immature myeloid cells suggests chronic
granulocytic leukaemia; cytogenetic and molecular studies the BCR–ABLfusion gene are indicated.

Lymphocytosis
Lymphocytosis is a feature of certain
infections, particularly infections
in children. It may be especially marked in pertussis, infectious mononucleosis, cytomegalovirus infection, infectious
hepatitis, tuberculosis, and
brucellosis. Lymphocytosis is also a common transient reaction to severe physical stress. Elderly patients with lymphoproliferative
disorders, including chronic
lymphocytic leukaemia and lymphomas, often present with lymphadenopathy and a lymphocytosis. Morphology and
immuno-phenotyping of the cells combined with histological examination of a bone marrow trephine
biopsy are used to classify these
disorders and to give an indi-cation of management and prognosis. It is
occasion-ally difficult to differentiate between a reactive and a neoplastic lymphocytosis. In this
situation, immunophenotyping,
immunophenotypic evidence of light chain restriction, and polymerase chain reaction for immunoglobulin or T-cell
receptor gene rearrangements may
indicate the presence of a monoclonal population of lymphocytes, thereby supporting a diagnosis of neoplastic,
rather than reactive,
lymphoproliferation. If lymph nodes are enlarged, a fine needle aspirate for cytology and immunocytochemistry or a lymph node biopsy
for histology and immunohistochemistry may be help-ful in diagnosis.

Monocytosis
A slight to moderate monocytosis may be
associated with some protozoal,
rickettsial, and bacterial infec-tions including malaria, typhus, and
tuberculosis.
High levels of monocytes in an elderly
patient suggest chronic myelomonocytic
leukaemia or, sometimes, atypical chronic
myeloid leukaemia. Because these
conditions fall into the myeloproliferative/myelodysplastic group of dis-orders, the diagnosis would
be supported by finding splenomegaly, quantitative and qualitative abnor-malities in other cell
lines, and a clonal cytogenetic abnormality.
Eosinophilia
Eosinophilia is typically associated with
allergic dis-orders including drug sensitivity, skin diseases, and parasitic infections. In most cases, the
cause is indi-cated from the clinical history, which should include details of all medications and foreign
travel, and by examination of the stool
and urine for parasites. A diagnosis of chronic eosinophilic leukaemia is made if there is an increase in blast
cells in the blood or marrow or if
there is cytogenetic or molecular evidence of an abnormal myeloid clone.

Idiopathic hypereosinophilic syndrome is
an unusual cause of
eosinophilia in which release of the contents of eosinophil granules results in damage to the heart, lungs, and other
tissues. This is a diagnosis of
exclusion, made only when detailed
investigations exclude all known causes.
It is necessary to
specifically exclude eosinophilic leukaemia and cytokine-induced eosinophilia result-ing from the presence of
a neoplastic clone of T cells before diagnosing a condition as the idiopathic hypereosinophilic syndrome.
Basophilia
Basophilia as an isolated finding is
unusual. How-ever, it is a common feature of myeloproliferative disorders and basophils may be
particularly pro-minent in chronic granulocytic leukaemia. In this condition, an increasing basophil count
may be the first indication of
transformation to a more aggressive course.
Thrombocytosis
Thrombocytosis is
often associated with infectious and inflammatory conditions such as
osteomyelitis and rheumatoid arthritis. Haematological causes of thrombocytosis
include chronic blood loss, red cell destruction, splenectomy, and rebound following
recovery from marrow suppression. Under these circumstances, a moderately
increased platelet count does not
usually have any
pathological
consequences. Primary (essential) thrombocythaemia belongs to the spectrum of myeloproliferative diseases and is characterized
by a persistently
high platelet count (often arbitrarily defined as greater than 600 ?10 /l) and thrombotic
or haemorrhagic
complications. Further investi-gations to confirm primary thrombocythaemia include bone marrow examination for increased
and abnormal
megakaryocytes and cytogenetic analysis.

Reductions in More Than
One Cell Line
A reduction in cell numbers occurs because
of in-creased destruction, reduced production, or increased pooling in the
spleen or other organ. Reduced production of cells may be the result of
aplastic anaemia, a lack of haematinics such as folate or vitamin B12, or
interference with normal haemopoiesis by infiltration (e.g., leukaemia,
lymphoma, multiple myeloma, metastatic carcinoma — often with
secondary myelofibrosis), infection (e.g.,
human immunodeficiency virus [HIV] infection, tuberculosis, leishmaniasis), or
exposure to toxins (e.g., alcohol) or myelosuppressive drugs (e.g.,
hydroxycarbamide* or busulphan). Certain myeloid neoplasms (e.g., “idiopathic”
myelofibrosis) and the myelodysplastic syndromes (MDS) are characterised by
cytopenias, and this is also sometimes a feature of acute myeloid leukaemia
(AML). A relatively common cause of a global reduction in circulating cells is pooling
of the cells in a grossly enlarged spleen
(hypersplenism), which may be secondary to
conditions such as myelofibrosis and portal hyper-tension. Examination of a
bone marrow aspirate and trephine biopsy specimen is often helpful in determining
the cause of bicytopenia or pancy-topenia for which no obvious cause can be
found.

Anaemia
There are many causes of anaemia, and a
logical classification would be according to mechanism:
·
Decreased production
·
Reduced lifespan of red cells
·
Blood loss
·
Splenic pooling

In practice, if the cause is not readily
apparent from the clinical circumstances and an automated blood count is
available, classification according to cell size is more practicable. The
choice of further investigations is then guided by the mean cell
volume (MCV) and red cell morphology in
addition to clinical features.

Anaemia is thus broadly divided into three
types:
·
Microcytic (low MCV)
·
Macrocytic (high MCV)
·
Normocytic (normal MCV)

Low MCV may be associated with low mean
cell haemoglobin (MCH). A low mean cell haemoglobin concentration (MCHC) is
less common and cor-relates with hypochromia. Examination of a blood film will
usually suggest the quickest route to the diagnosis; confirmation may require
the more specific tests, which are given in the text. The presence of
basophilic stippling in a patient with microcytic red cells suggests thalassaemia
trait or lead poisoning. A dimorphic blood film is typical of congenital
sideroblastic anaemia but is more often the result of iron defi-ciency responding
to treatment. Pappenheimer bodies suggest that a microcytic anaemia is the
result of sideroblastic erythropoiesis.

Microcytic Anaemia
The most common cause of anaemia worldwide
is iron deficiency. It can be suspected from a low MCV and the presence of
hypochromic, microcytic red cells. Laboratory confirmation of iron deficiency
may include measurements of serum ferritin, serum iron plus either total
iron-binding capacity or transferrin assay, red cell proto-porphyrin, and
staining of bone marrow aspirates for iron. A diagnosis of iron deficiency requires
a search for the cause. This should include
specific questions relating to blood loss
and dietary insufficiency and may require stool examination for parasites and
occult blood, endoscopic examination of the gastrointestinal tract to exclude
occult malig-nancy, and serological or other tests for coeliac disease. The
differential diagnosis of iron deficiency anaemia includes anaemia of chronic
disease. Clinical and laboratory features of inflammation may suggest this
diagnosis, which is confirmed by demonstration of normal or high serum
ferritin, low serum iron, and low transferrin and iron binding capacity.
The thalassaemias also cause microcytosis,
but both α and β thalassaemia are usually associated with an
increased red blood cell count and a normal or near-normal Hb despite a
considerable reduction of the MCV and MCH, whereas in iron deficiency the MCV
and MCH do not fall until the Hb is significantly reduced. Further
investigations, such as haemoglobin electrophoresis, high-performance liquid
chromato-graphy (HPLC), or measurement of HbA2 and HbF
usually confirm the diagnosis of β
thalassaemia trait. The diagnosis of α thalassaemia trait is more difficult;
detection of infrequent HbH inclusions is usually possible in α thalassaemia
trait, but definitive diagnosis requires DNA analysis. The diagnosis of α +
thalssaemia trait is of less clinical importance; HbH inclusions may not be
detected, so DNA analysis is needed.
Macrocytic Anaemia
A high MCV with oval macrocytes and
hyper-segmented neutrophils suggests folate or vitamin B12 deficiency and is an
indication for assays of these vitamins (see Chapter 8); subsequent
investi-gations could include malabsorption studies,
serological test for coeliac disease, and
either tests for intrinsic factor antibodies or a Schilling test to detect
pernicious anaemia. If intrinsic factor antibodies are detected, a Schilling
test is not necessary. A high MCV may also be associated with alcohol excess
and liver disease or drugs such as hydroxycarbamide or zidovudine. Macrocytosis
resulting from chronic haemolysis is associated with
increased numbers of immature red cells,
which appear slightly larger and more blue than normal red cells on a
Romanowsky-stained peripheral blood film. Supravital staining of blood films
(p. 40) or an automated reticulocyte count can be used to confirm reticulocytosis.
Untreated anaemia associated with polychromasia is likely to indicate blood
loss or haemolysis. The combination of red cell fragments, thrombocytopenia,
and polychromasia indicates microangiopathic haemolytic anaemia and should trigger
further tests such as a platelet count, coagulation studies, assessment of
renal function, and a search for infection or neoplastic disease. This further
assessment is urgent because these may be features of thrombotic
thrombocytopenic purpura, which requires speedy treatment by plasma exchange.
Normocytic Anaemia
Normochromic, normocytic anaemia is
frequently the result of an underlying chronic, non-haematological disease.
Investigations should include screening for renal insufficiency, subclinical infections,
autoimmune diseases, and neoplasia. In
the presence of anaemia, a lack of
polychromasia, confirmed by reticulocytopenia, points toward a primary failure
of erythropoiesis or blood loss or haemolysis without compensatory red cell
produc-tion. Examination of the bone marrow may be help-ful in demonstrating
haematological causes for the normochromic, normocytic anaemia such as aplastic
anaemia or early myelodysplastic syndrome. Stain-ing for iron may also show
that there is a block in iron metabolism suggestive of anaemia associated with
chronic inflammatory disease.
Leucopenia
Neutropenia
Once physiological variation, ethnicity,
and familial or cyclic neutropenia have been excluded, the non-haematological
causes of isolated neu-tropenia to be considered include overwhelming infection,
autoimmune disorders such as systemic lupus erythematosus, irradiation, drugs
(particularly anticancer agents), and large granular lymphocyte leukaemia. Bone
marrow examination may assist in determining whether the problem is the result
of peripheral destruction (increased marrow myeloid precursors) or stem cell
failure (lack of narrow myeloid precursors). Typical marrow appearances occur
in drug-induced neutropenia, in which there is a relative paucity of mature
neutrophils and in Kostmann’s syndrome (infant genetic agranulocytosis) where
there is maturation arrest at the promyelocytic stage.

Reduced Numbers of Lymphocytes, Monocytes,
Eosinophils, and Basophils Lymphocytes, eosinophils, and basophils may all be reduced
by stress such as surgery, trauma, and infec-tion. Lymphopenia with
neutrophilia is a common combination of haematological abnormalities in severe
acute respiratory syndrome. Lymphopenia, especially affecting the CD4 cells,
may occur in HIV infection and renal failure. Monocytopenia is typically found
in hairy cell leukaemia, which is also associated with pancytopenia, typical bone
marrow histology, and lymphocytes with a characteristic cytology and
immunophenotype.
Thrombocytopenia
Thrombocytopenia is a common isolated
finding, and it is important to ensure that the laboratory result reflects a
true reduction in platelet count before embarking on further diagnostic tests.
Frequent causes of spurious thrombocytopenia include blood clots in the sample,
platelet aggregation, and platelet satellitism. Platelet aggregation, which can
be seen on the blood film, may occur in vitroas theresult of a
temperature-dependent or anticoagulant-dependent autoantibody. Small platelet
aggregates are also seen in slides that have been made directly from a
fingerprick sample. True thrombocytopenia is most frequently the result of
anticancer chemo-therapy, HIV infection, autoantibodies (“idiopathic” autoimmune
thrombocytopenic purpura), other drugs (such as thiazide diuretics), alcohol
excess, hypersplenism, and MDS (in the elderly). The clinical circumstances,
together with bone marrow examination and relevant serological tests, should enable
these conditions to be differentiated. Throm-bocytopenia associated with other
complications, such as thromboses, disturbed renal or hepatic function, and
haemolytic anaemia, should prompt investigations for other diseases such as
thrombotic thrombocytopenic purpura or the HELLP (Haemolysis + Elevated Liver
enzymes + Low Platelet count) syndrome. A bone marrow examination is often carried
out early in the investigation of throm-bocytopenia because it is helpful in
excluding con-ditions such as acute leukaemia, which occasionally present with
isolated thrombocytopenia.
Pancytopenia
Pancytopenia (reduction in the white cell
count, Hb, and platelet count) is most often the result of anticancer
chemotherapy, HIV infection, hyper-splenism, and bone marrow infiltration or
failure. Careful examination of a blood film is important if the reason for the
pancytopenia is not apparent from the clinical history. If this does not reveal
the cause, bone marrow aspiration and trephine biopsy may be needed.
Qualitative Abnormalities of Blood Cells
In health, only the most mature forms of
cells appear in the peripheral blood. Earlier, less mature cells, such as nucleated red blood cells,
polychromatic red cells, myelocytes, and metamyelocytes, may be released from
the bone marrow in conditions where the bone marrow is overactive (e.g., acute
haemolytic states or recovery after suppression) or functionally abnormal.
Their presence in the peripheral blood indicates that active haemopoiesis is
taking place.
Abnormalities of All Cell Lines
The combination of anisopoikilocytosis,
mild macrocytosis, hypogranular neutrophils with abnor-mal nuclear morphology,
and platelet anisocytosis, often with quantitative abnormalities, is virtually pathognomonic
of a myelodysplastic syndrome. These features are reflected in the bone marrow
with disturbance of the normal developmental pathway and nuclear:cytoplasmic
asynchrony. Cytogenetic
studies can confirm the diagnosis when
cytological abnormalities are minor and also can assist in determining the
prognosis.
Abnormalities of Individual Cell Lines
Red Cells
Congenital abnormalities of the red cell
affecting the structure (e.g., spherocytosis, elliptocytosis) and content
(e.g., haemoglobinopathies, enzymopathies) often produce typical morphological
changes. The type of changes will guide further
investigations toward analysis of
structural proteins, haemoglobin electrophoresis, or HPLC and enzyme assays.
Acquired red cell abnormalities may also help to indicate underlying pathology.
For example, target cells may prompt investigation of liver function, whereas
rouleaux may indicate the need for investigations for multiple myeloma or inflammatory
conditions such as rheumatoid arthritis.
White Cells
Congenital abnormalities of neutrophils
are unusual, but similar morphological abnormalities (e.g., Pelger–Hu?t cells)
may be seen in acquired con-ditions such as myelodysplastic syndrome. Reactive changes
in lymphocytes including basophilic, faceted cytoplasm, are typically seen in
infectious mononucleosis, which can be diagnosed using an appropriate screening
test or, if this is negative, by demonstration of immunoglobulin M (IgM)
antibodies to the Epstein–Barr virus. These atypical lymphocytes can sometimes
be difficult to differentiate from circulating lymphoma cells. Bone marrow
histology, combined with immunopheno-typing studies and determination of
lymphocyte clonality by demonstration of light chain restriction or by gene
rearrangement studies, may be needed to reach a firm conclusion.
Platelets
Platelets that function poorly may not
necessarily appear morphologically abnormal, although some-times they are
hypogranular or larger than normal. A normal platelet count with a prolonged
bleeding time is characteristic of a disorder of platelet func-tion, but some
patients with abnormal platelet function are also thrombocytopenic. Hereditary
dis-orders of platelet function are uncommon and
usually present as a bleeding diathesis.
When a qualitative disorder of platelets is suspected, platelets should be
examined to assess size and to detect the cytological features of the grey
platelet syndrome. They can broadly be divided into two categories:
abnormalities of the platelet membrane (e.g., Bernard–Soulier syndrome,
Glanzmann’s thrombasthenia) and of platelet secretory function (e.g., storage
pool diseases). In comparison, acquired disorders of platelet function are
common.
Haematological conditions associated with
platelet dysfunction include myeloproliferative and myelo-dysplastic disorders
and dysproteinaemias. Many
widely prescribed drugs can interfere with
platelet function, including aspirin and non-steroidal anti-inflammatory
agents, whereas systemic conditions, particularly chronic renal failure and
cardiopulmonary bypass, are associated with a bleeding tendency as a result of
qualitative platelet defects. Most of these acquired functional defects are not
associated with any abnormality in platelet appearance but in the myelodysplastic
and, to a lesser extent, in the myelo-proliferative disorders there may be
hypogranular and giant platelets.
SPECIFIC TESTS FOR COMMON
HAEMATOLOGICAL DISORDERS
Common haematological disorders are
outlined in the following sections with suggestions for investi-gations that
may be helpful in confirming the diagnosis. The lists are not intended to be
exhaustive because the range of tests provided locally will depend on the
availability of expertise and tech-nology. The investigations discussed are those
that are likely to be available within a general
haematology department.
Red Cell Disorders
Microcytic Hypochromic Anaemias
For more information, see Chapters 7 and
12.
• Measurement of serum ferritin or iron
plus either total iron-binding capacity or transferrin assay, red cell
protoporphyrin
• Bone marrow aspirate with staining for
iron
• Stool examination for occult blood;
blood loss studies with 51
Cr-labelled red cells
• Tests for malabsorption
• Serological tests for coeliac disease
• Endoscopic examination with biopsy
• Serum lead (if lead poisoning is
suspected)
If thalassaemia is suspected:
• Haemoglobin electrophoresis plus Hb A2 and
Hb F measurements or HPLC
• Haemoglobin H preparation
• Family studies
• DNA analysis
Macrocytic Anaemias
Where macrocytic, megaloblastic erythroid
maturation is demonstrated, further investigations should be undertaken as
described in Chapter 8. If the blood film is typical of megaloblastic anaemia,
relevant assays and further investigations are often per-formed without a bone
marrow aspirate being done.
Macrocytosis may also be secondary to
common conditions such as alcohol excess, liver disease, myelodysplastic
syndrome, hydroxycarbamide administration, and hypothyroidism. Reticulocytosis from
any cause can also increase the MCV.
Aplastic Anaemia
• Bone marrow aspirate and trephine biopsy
• Acidified serum (Ham’s) test for
paroxysmal nocturnal haemoglobinuria (urine examination for haemosiderin and
neutrophil alkaline phosphatase if Ham’s test is positive)
• Vitamin B12 and folate assays
• Viral studies, particularly for
Epstein–Barr and hepatitis viruses.
If Fanconi’s anaemia is suspected:
• Studies of sensitivity of chromosomes to
breakage
by DNA crosslinking agent
• Radiology of hands and forearms
Haemolytic Anaemias
A haemolytic process may be suspected by
the presence of a falling haemoglobin, a reticulocytosis, and jaundice with an
increase in unconjugated bilirubin level.
The blood may appear entirely normal in
some patients with white cell disorders (e.g., lymphoma, myelomatosis, immune
deficiency, neutrophil dys-function). Changes in white cell numbers or morphology
may occur rapidly in response to local or systemic disorders. The investigation
of white cell disorders is more likely to require marrow examination than
investigation of red cell disorders, especially when a primary marrow disorder
is suspected. In chronic leukaemias, bone marrow examination may add little to
the diagnosis, but the pattern of infiltration may have prognostic significance,
e.g., in chronic lymphocytic leukaemia.
The distribution of white cells is better
appreciated in trephine biopsies, whichare particularly important in lymphomas.
Acute Leukaemia
• Bone marrow aspirate
• Bone marrow trephine biopsy if an adequate
bone marrow aspirate is not obtained
• Cytochemical stains
• Blood or marrow immunophenotyping,
unless obviously myeloid
• Cytogenetic analysis
• Molecular studies (e.g., fluorescence in
situ hybridization) for rearrangements of specific oncogenes
Neutropenia
• Serial neutrophil counts for cyclical
neutropenia
• Tests for antineutrophil antibodies
• Bone marrow aspirate and trephine biopsy
• Autoantibody screen and investigations
for systemic lupus erythematosus
• Vitamin B12 and folate assays
• Acidified serum (Ham’s) test
Chronic Granulocytic
Leukaemia
• Bone marrow aspirate
• Cytogenetic analysis
• Molecular studies (e.g., fluorescence in
situ hybridization) for BCR–ABLrearrangement
• Neutrophil alkaline phosphatase score,
if cyto-genetic and molecular genetic analysis are not available
Chronic
Lymphoproliferative Disorders/ Lymphadenopathy
• Serological screening for infectious
mononucleosis, cytomegalovirus infection, HIV infection, and toxoplasmosis (if
infectious cause suspected)
• Bone marrow aspirate and trephine biopsy
(for detection of the presence and distribution of abnormal lymphocytes)
• Immunophenotyping
• Serum protein electrophoresis and
immuno-globulin concentrations
• Serum urate, calcium, and lactate
dehydrogenase (LDH)
• Lymph node biopsy (aspiration or
surgical)
• Cytogenetic or molecular genetic
analysis including investigation for immunoglobulin heavy chain or T-cell
receptor gene rearrangement if the diagnosis of lymphoma is in doubt
• Radiological studies (X-ray,
ultrasonography, computed tomography scan, magnetic resonance imaging)
Myelomatosis
• Bone marrow aspirate
• Bone marrow trephine biopsy if a
cellular aspirate is not obtained
• Serum protein electrophoresis and
immunoglobulin concentrations
Serum albumin and calcium measurements
• β2-microglobulin
• Urine (random and 24 hours) for
Bence–Jones protein detection and quantitation
• Tests of renal function
• Radiological skeletal survey
• Serum free light chain quantification
and ratio
Other Disorders
Myeloproliferative Disorders
• Blood volume, red cell mass, and plasma
volume (for polycythaemia) measurements
• Bone marrow aspirate and trephine biopsy
• Arterial oxygen saturation and
carboxyhaemo-globin level
• Abdominal ultrasound examination
• Neutrophil alkaline phosphatase
• Vitamin B12 (or B 12 -binding capacity)
• Serum urate
• JAK2 analysis
Myelodysplasia
• Bone marrow aspirate and trephine biopsy
• Cytogenetic analysis “Idiopathic”
Myelofibrosis
• Bone marrow trephine biopsy
• Red cell folate assay
• Urate
If splenectomy is
contemplated:
• Ferrokinetic and red cell survival
studies
• Spleen scan and red cell pool
measurement.
Pancytopenia with
Splenomegaly
• Bone marrow aspirate and trephine biopsy
• Bacterial culture of marrow for
tuberculosis
• Marrow examination for amastigotes of
Leishmania donovani
• Biopsy of palpable lymph nodes
(aspiration or surgical)
• Vitamin B12 and folate assays
• Liver biopsy
• Splenic aspirate
• Acidified serum (Ham’s) test
• Serum rheumatoid factor and autoantibody
screen
• Laparotomy and splenectomy.
The rationale behind these tests, and
details of investigations outside general haematology practice, can be found in
comprehensive haema-tology textbooks, in electronic databases.
Since the late 1970s, acute leukaemia and
the MDS have usually been defined and further classified according to the
proposals of the French–American–British (FAB) group. These classifications are
now being supplanted by the World Health
Organization (WHO) classifications,
published in definitive form in 2001. The WHO classifications are much broader
in their aims and include myelo-proliferative and chronic lymphoproliferative disorders.
Application of the WHO criteria requires
the results of immunophenotyping and cytogenetic analysis. The FAB
classifications therefore continue to have a place (a) when these techniques
are not available and (b) in making a provisional morpho-logical diagnosis
while awaiting the results of further tests. It may be necessary to issue a provisonal
report, so that treatment can commence, with a supplementary report following
when all diagnostic procedures have been completed. It is important that,
whichever classification is used, the criteria are strictly observed so that
there is consistency between different centres and countries. To avoid any
possibility of confusion, FAB terminology (e.g., M1, M2) should not be applied
if the WHO classsification is being used.
Classification of Acute
Myeloid Leukaemia
The FAB criteria for a diagnosis of AML
are as follows:
1.Blast cells must constitute at least 30%
of all bone marrow cells or
2.When erythroid cells are at least 50% of
bone marrow cells and blasts cells must be at least 30% of non-erythroid cells
(lymphocytes, plasma cells, macrophages, and mast cells also being exluded from
the count) or
3. The characteristic cytological features
of acute hypergranular promyelocytic leukaemia or
4. Blast cells are shown to be myeloid by
either there being at least 3% of blast cells positive for Sudan black B,
myeloperoxidase, or nonspecific esterase or by demonstration of myeloid
antigens on immunophenotyping.
The WHO classification categorises cases
as AML if the following criteria are met:
1.There are at least 20% of blast cells of
myeloid lineage in the blood or bone marrow or
2.If the erythroid cells are at least 50%
of bone marrow cells, blast cells are at least 20% of nonerythroid cells, or
3.Primitive erythroid cells constitute at
least 80% of bone marrow cells or
4.There is a myeloid sarcoma (granulocytic
sarcoma) or
5.One of a number of specified chromosomal
rearrangements is present.
It should be noted that the WHO
classification is hierachical. If appropriate, cases are first assigned to the
category of therapy-related leukaemia. Next, cases are assigned, if
appropriate, to the category of AML with recurrent genetic abnormalities. Cases
continue to be assigned to successive categories in the order with remaining
cases finally being categorised as “AML not otherwise categorized.” This final
group is further subdivided into categories resembling those of the FAB classification
(but defined in quite a different manner). The “not-otherwise-categorized”
group includes several entities that are either newly defined (acute
panmyelosis with myelofibrosis and pure erythroid leukaemia) or were not
specifically men-tioned in the FAB classification (acute basophilic leukaemia
and myeloid sarcoma.
The FAB criteria for a diagnosis of MDS
are that there is evidence for a myeloid neoplasm with ineffective haemopoiesis
but the criteria for AML are not met. Blast cells must be less than 30% in the bone
marrow. In the WHO classification, there must be evidence for a myeloid
neoplasm with ineffective haemopoiesis and blasts must be less than 20% in both
blood and bone marrow. In additon, the specific cytogenetic abnormalities must
be absent (or the case is categorized as AML, regardless of the blast count).
There is a further major difference between the two classifications, specifically
that chronic myelomonocytic leukaemia is classified as MDS in the FAB
classification, whereas in the WHO classification it is assigned to a new category
of disorder designated myelodysplastic/myeloproliferative diseases. It will be noted
that cytogenetic analysis is essential for the application of the WHO
classification because cases
of the 5q- syndrome cannot otherwise be
recognized.
Like the WHO classification of AML, this
is a hierachical classification. Therapy-related MDS is categorized with
therapy-related AML. Remaining cases are then assessed as to whether they meet
the criteria for the 5q- syndrome. If they do not, they are assigned to the
remaining categories, depending on the number of lineages showing dyplasia, the
percentage of ring sideroblasts, the presence or
absence of Auer rods, and the percentage
of blast cells in the blood and marrow. The details of the WHO classification
of the myelodysplastic/ myeloproliferative diseases.
Both the FAB and WHO classifications
require that an acute leukaemia be positively shown to be lymphoid before it is
categorized as acute lympho-blastic leukaemia (ALL) so as to avoid
inadvertently categorising FAB M0 and M7 AML as ALL. The WHO classification
groups together ALL and lymphoblastic lymphoma, using the designations
precursor B lymphoblastic leukaemia/lymphoblastic lymphoma and precursor T
lymphoblastic leukaemia/lymph-oblastic lymphoma. These designations are clearly
too cumbersome to use in clinical practice, and undoubtedly haematologists will
continue to refer to “acute lymphoblastic leukaemia.” The FAB group classified
ALL into three morphological categories, designated L1, L2, and L3 ALL. It is
of little significance whether a case falls into the L1 or L2 category, and this
distinction can be dropped.
However, L3 morphology—the presence of
“blast cells” with basophilic cytoplasm and vacuolation—is of considerable
clinical significance. In most, but
not all, of these cases the cells are
immunologically mature, expressing surface membrane immunoglobulin, and the
condition represents a leukaemic presentation of Burkitt’s lymphoma. The WHO classification
categorizes such cases as Burkitt’s lymphoma. This is more appropriate than
their being categorized as ALL because the treatment of these cases differs
very considerably from the treatment of ALL.
The WHO classification of the
myeloproliferative disorders is summarized. Most of these conditions are
defined in accordance with established haematological practice. However, the
method of distinguishing between essential thrombocythaemia and idiopathic
myelofibrosis differs from previous practice with many cases that would
previously have been categorized as essential thrombocythaemia now being
classified as the cellular phase of myelo-fibrosis; whether this
classsification will be widely accepted remains to be seen. The WHO
classification of myeloproliferative disorders takes little account of
cytogenetic or molecular genetic analysis, only Ph-positive chronic myeloid
leukaemia being defined on this basis. It should be noted that the WHO classification
defines a case with hypereosinophilia as eosinophilic leukaemia, rather than as
the idiopathic
hypereosinophilic syndrome, when there is
evidence of clonality. The 4q12 syndrome, resulting from a cryptic deletion at
4q12 with formation of a FIP1L1-PDGFRA fusion gene, is therefore categorized as
eosinophilic leukaemia.
Leukocyte Disorders
The importance of
an intact host immunoinflammatory response in maintaining periodontal health
underscores the changes that may occur when patients have leukocyte disorders
(see picture below). The neutrophil or polymorphonuclear leukocyte is the first
line of defense against periodontal pathogens. A defect in this cell line has
clear negative consequences, often resulting in severe periodontal destruction
at an early age.

Although the clinician will not encounter
these disorders on a routine basis, the severe periodontal destruction
associated with neutrophils disorders can be overwhelming for both the patient
and the provider.
Neutrophils
circulate within the bloodstream and must pass out of the vessels and into the
tissues to defend against periodontal pathogens. Pathogens and host cells
produce chemotactic agents that signal neutrophils to enter an area of
infection. To be fully functional, neutrophils must be able to do the following:
1. Slow down their flow within the vasculature--a process
involving cell surface glycoproteins known as selectins on the surface of
neutrophils and endothelial cells lining the blood vessels. Up-regulation of
these selectins results in "rolling" of the
neutrophils along the endothelial lining of the vessel.
2. Adhere to the endothelial cells—a process involving interaction between
receptors called integrins on the surface of neutrophils and receptors on the
endothelial cell surface.
3. Pass between the intercellular spaces of the endothelial lining to exit
the vessel and to enter the perivascular tissues—a process known
as diapedesis.
4. Move toward the pathogens that they will attack—a process
involving locomotion (movement) of the neutrophil toward the bacteria or host
cells that are producing factors called chemoattractants. This is the process
of chemotaxis (directed movement toward a chemoattractant).
5. Bind to the pathogens, engulf them, and move them into the intracellular
environment of the neutrophil—a process known as phagocytosis.
6. Kill the offending pathogen—a process called degranulation, involving
enzyme release from intracellular granules. Another method of killing involves
release of free oxygen radicals. This process not only kills the offending
pathogen but may result in host tissue damage, especially if the response is
exuberant.

Fig.1(A,B,C).
A, Soft tissue swelling associated with teeth #17
and #18 in a 22-year-old man. Teeth were vital. B, Radiograph revealed periradicular radiolucency
associated with the mesial root of #17, with loss of trabeculation in the
alveolar crest distal to #17 and between #17 and #18. C, Biopsy revealed numerous giant cells in fibrous
stroma, features of a classic giant cell granuloma. Systemic work-up disclosed
hyperparathyroid state.
Defects in the
response of neutrophils to pathogens can occur at any step along this pathway. This results in an inability to clear a bacterial infection and allows
bacteria and their products to continue to destroy host tissues.
Leukocyte Adhesion Deficiency
Leukocyte adhesion deficiency (LAD) is an inherited disorder that follows
an autosomal recessive pattern. There have been just more than 600 cases
described, each identified shortly after birth. More than 75% of children will
die before the age of 5 years if they do not receive a bone marrow transplant.
Leukocyte adhesion deficiency is caused by a deficiency in cell surface
integrins that prevents the neutrophil from adhering to the vessel wall at the
site of an infection. Neutrophils are
unable to migrate into the affected tissues and remain within the vasculature.
This prevents them from attacking bacterial pathogens. Patients have early loss of teeth, severe
alveolar bone loss and attachment loss, and severely inflamed gingival tissues,
often with ulceration and necrosis. Both primary and permanent teeth are affected. Treatment is difficult, involving mechanical
debridement, topical antimicrobials, and systemic antibiotics. Unfortunately,
treatment rarely results in long-term retention of teeth (see picture below).

Fig.2 Patient with generalized aggressive periodontitis.
A, Clinical appearance of a 12-year-old boy
with generalized aggressive periodontitis resulting from leukocyte adhesion
deficiency (LAD).
B, Gingival tissues are fiery
red, painful to touch, and bleed profusely on stimulation.
C, Panoramic radiograph reveals severe bone loss at
age 12 years affecting all permanent teeth. Teeth #23 and #26 exfoliated
spontaneously.
Neutropenia
The normal adult
absolute neutrophil count (ANC) is between 1800 to 8000 cells/^l. Neutropenia
(low ANC) is considered clinically significant when the ANC decreases to less
than 1000 cells/^l. Chronic neutropenia is defined as a low ANC for greater
than 6 months. The risk for infection caused by neutropenia is inversely proportional
to the ANC. When the ANC is less than 500 cells/^l, control of endogenous
microbiota is often impaired and the risk for serious infection increases. An
ANC less than 200 cells/^l results in an inability to mount an inflammatory
response.
Chronic benign neutropenia
Chronic benign neutropenia (CBN) is characterized by a prolonged noncyclic
neutropenia as the sole abnormality. The neutropenia is not associated with any
underlying disease. CBN is the most common form of neutropenia in
infants and children younger than 4 years. The clinical presentation is
variable, ranging from benign to life-threatening; however, most people with
CBN live a normal lifespan. An increased incidence of recurrent oral
ulcerations, upper respiratory infections, otitis media, cellulitis,
lymphadenopathy, pneumonia, and sepsis occurs as a result of the decreased
neutrophil response. The risk for infection appears to decrease with age. Oral manifestations of CBN may include
hyperplastic, edematous, and fiery red gingiva with areas of desquamation,
although not all patients with CBN are similarly affected. Severe pocketing and
bone loss may occur. Ulceration, chronic gingivitis, and chronic periodontitis
also have been reported. Within the periodontal tissues, the chronic lack of neutrophils may be
counterbalanced by increased antibacterial activity from monocytes. This may
explain the milder periodontal findings in some patients with CBN.
Cyclic neutropenia
Cyclic neutropenia is characterized by periodic recurring symptoms
of fever, malaise, mucosal ulcers, and possibly life-threatening infections
related to cyclical fluctuations in the number of neutrophils. This disorder usually presents before age 10 years with episodes of fever,
malaise, mood swings, and oral ulcerations that can last 3 to 6 days and recur
approximately every 3 weeks. The interval between neutropenic episodes is not
always clinically evident and may require frequent laboratory studies to
identify. A complete blood count performed twice weekly for 6 weeks generally
provides an accurate picture of the cycle. For most patients, the cycle is
approximately 21 days, with a 3- to 10-day period of severe neutropenia.
Cyclic neutropenia
tends to improve with age. Its clinical presentation may vary widely among individuals.
Although usually not fatal, death can occur due to pneumonia, cellulitis,
gangrene, or peritonitis. Oral conditions
associated with cyclic neutropenia may include recurrent severe gingivitis and
oral ulcerations. Periodontitis may progress more rapidly than expected because
of periodic diminishment of the neutrophil response. Unfortunately, even with
the best of professional and home care, teeth are often lost because of
advancing periodontal disease. Treatment to increase neutrophil levels has been
successful using recombinant human granulocyte colony-stimulating factor
(G-CSF) given three times per week. G-CSF is a hematopoietic growth factor that
simulates the proliferation and differentiation of neutrophils. It has been
widely successful in correcting chemotherapy-induced neutropenia in patients
with cancer, greatly decreasing the risk for life-threatening infections during
periods of immunosuppression.
Congenital neutropenia
Congenital neutropenia, also known as Kostmann
syndrome, is an inherited disorder manifesting in infancy and characterized by
severe bacterial infections. Diminished ANC is the result of arrested
neutrophil hematopoiesis. Oral symptoms are virtually universal in congenital
neutropenia. Despite their young age, patient with this syndrome not only
demonstrate severe gingivitis, but most also have periodontitis with
significant alveolar bone loss. In the past, most patients with congenital neutropenia died within the
first year of life; however, aggressive antibiotic therapy has more recently
prolonged the lifespan of these children.Congenital
neutropenia is now treated with G-CSF, which is effective at increasing the ANC
to more than 1000/^l in most patients. Although G-CSF treatment improves
symptoms, it is not curative and most patients demonstrate cyclic improvements
followed by relapses in neutrophil levels. Even with G-CSF treatment, most of
these patients have persistent gingivitis, which tends to wax and wane
depending on their ANC.
Agranulocytosis
Agranulocytosis is characterized by a reduction or complete
elimination of granular leukocytes (neutrophils, basophils, eosinophils). The
decreased number of granulocytes can result from either a decreased production
or an increased peripheral destruction of cells. Decreased production of
granulocytes is often caused by bone marrow hypoplasia; however, it can also be
the result of an idiosyncratic drug reaction. Patients with agranulocytosis are often febrile
and may exhibit necrotizing nonpurulent lesions of mucous membranes, including
oral, gastrointestinal, and vaginal membranes. Oral signs and symptoms include generalized,
painful stomatitis, spontaneous bleeding, and necrotic tissue. Severe
gingivitis, rapidly progressive bone loss, and tooth loss may appear at an early
age. Cases related to drug idiosyncrasy usually occur in adulthood.
Lazy leukocyte syndrome
Lazy leukocyte syndrome is a rare disorder characterized by quantitative
and qualitative neutrophil defects. Deficiency in neutrophil chemotaxis
combined with systemic neutropenia results in recurrent infections. Impaired
neutrophil motility inhibits their migration into tissue sites of inflammation.
In the few reported cases of lazy leukocyte syndrome, all have had oral
manifestations. In addition to systemic
signs and symptoms such as high fever, cough, pneumonia, and purulent skin
abscesses, oral manifestations include painful stomatitis, gingivitis,
recurrent ulcerations of the buccal mucosa and tongue, rapidly progressive bone
loss, and tooth loss at an early age.
Papillon-Lefevre Syndrome


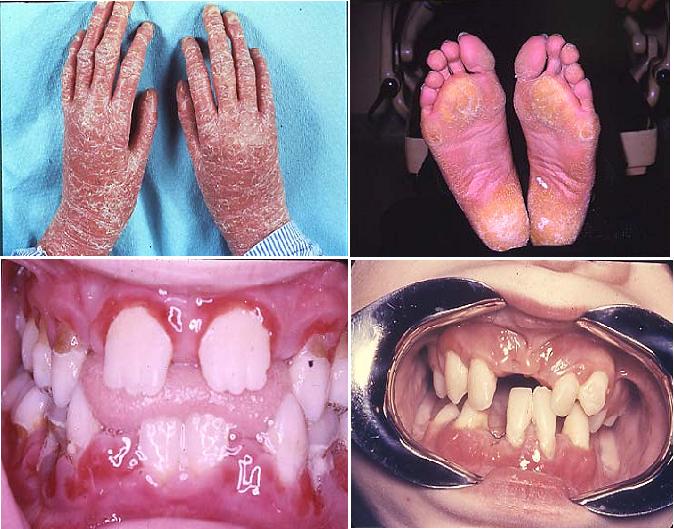{kind=link}
{kind=link}
Papillon-Lefevre syndrome (PLS) belongs to a heterogenous group of 19
different skin diseases characterized by hyperkeratosis of the palms of the
hands and soles of the feet (palmar-plantar hyperkeratosis). PLS is caused by mutations in the cathepsin C
gene located on chromosome 11. Cathepsin C is a protease, normally found in
high levels in epithelium and immune cells such as neutrophils, which acts to
degrade proteins and activate proenzymes in immune cells. Patients with PLS
have little or no cathepsin C activity.
PLS differs from
other members of this group of hyperkeratoses in that patients with PLS
universally have generalized rapid destruction of the periodontal attachment
apparatus resulting in premature loss of primary and permanent teeth. The
presence of neutrophil defects in PLS is commonly noted. Diminished chemotaxis,
phagocytosis, and intracellular killing of certain bacteria have been reported
in some but not all cases. It is possible that neutrophil defects are not
entirely responsible for the findings in PLS. Some authors have hypothesized
that the hereditary defect in PLS is located in the epithelial barrier, which
in the gingival sulcus may lead to a reduced defense against pathogenic bacteria. Alterations in cementum, collagenolytic activity in
the periodontal ligament, and osteoclastic activity have also been suggested in
some patients with PLS. Taken together, these findings could explain the
aggressive periodontal destruction seen in patients with PLS even in the
absence of significant neutrophil abnormalities.
The periodontal
condition in PLS is difficult to treat, and use of conventional mechanical
debridement rarely has been successful. Systemic administration of synthetic
retinoids, when combined with meticulous plaque control, debridement, topical
antimicrobials such as chlorhexidine, and systemic antibiotic therapy, may give
the best chance for preventing progression of periodontitis.
Treatment Retinoid therapy: Improves the skin condition but not
the periodontal therapy. Periodontal condition: No effective treatment
Leukemias
Leukemia is a
neoplastic disorder of the blood-forming tissues, primarily affecting
leukocytes. This heterogenous group of diseases arises from a neoplastic
proliferation in the bone marrow. The replacement of normal bone marrow
elements by leukemic cells causes decreased production of erythrocytes, normal
white blood cells, and platelets. The clinical result is anemia, with weakness,
fatigue, pallor of skin, and mucous membranes; thrombocytopenia with associated
bleeding tendencies; and leukopenias resulting in increased susceptibility to
infection. Leukemias are classified as either acute or chronic, depending on
the presentation of the disease. They are further classified relative to the
predominant cell affected as either lymphocytic or myelocytic. Monocytic leukemias form a subgroup of myelocytic leukemia.
Oral involvement is common in leukemia and may represent the first sign of
the disease. Dental professionals were responsible for initiating the diagnosis
of leukemia in 25% to 33% of cases. Overall, 15% to 80% of patients with leukemia have oral
manifestations, with the acute forms presenting oral signs in approximately 65% of cases, compared with only 30% in chronic leukemias.
Oral petechiae or bleeding, mucosal ulceration, and gingival enlargement are
the most common signs. Acute periodontal infection, pain, pharyngitis, and lymphadenopathy also may
be seen.
Gingival enlargement may be localized or generalized and represents an
infiltration of leukemic cells into the gingiva, and less frequently into bone (Fig. 3). Gingival enlargement is most common in acute
monocytic leukemia (67% of cases), followed by acute myelomonocytic
leukemia (18.5%), and acute myelocytic leukemia (4%).— The enlarged gingiva tends to be relatively firm
in texture and most prominent in the interdental regions. The marginal tissues
may be bluish-red or cyanotic. Gingival enlargement creates pseudopockets where
plaque accumulates, stimulating a host response that may further exacerbate the
swelling. Gingival bleeding is also common, and may be an early indicator of
leukemia. Oral mucosal ulcers are a frequent finding in patients with leukemia.
These lesions may result from bacterial invasion caused by severe leukopenia or
from mucosal atrophy caused by a direct effect on epithelial cells of the
chemotherapeutic drugs used to treat leukemia. Trauma from a dental prosthesis
or teeth may result in large secondarily infected ulcers progressing to facial
cellulitis and septicemia.

Figure 3.Leukemic gingival enlargement in 33-year-old man.
Biopsy of gingiva revealed large leukemic infiltrate.
Treatment for
leukemia may include chemotherapy, radiation therapy, and BMT, each of which
has the potential to produce a wide range of oral complications. Mucositis,
xerostomia, and secondary infection with a variety of bacterial, viral, and
fungal agents may occur. Candidiasis is
almost universally seen in hospitalized patients with leukemia undergoing
chemotherapy. Infections with unusual organisms (e.g., Pseudomonas and Klebsiella species) are
common in this group of patients. Many drugs used for chemotherapy are
neurotoxic and may cause intense oral pain, which is usually transient. These
symptoms must be distinguished from pain of odontogenic origin. Patients
undergoing BMT require special consideration because they receive very
high-dose chemotherapy, often in combination with total body irradiation. The
extreme immunosuppression experienced by patients with BMT predisposes to
systemic spread of even mild infections. A large percentage of patients with
BMT develop graft-versus-host disease, a condition where transplanted
immunocompetent marrow cells recognize the host tissues as foreign and react
against them, resulting in fever, mucosal ulcerations, skin erythema, and
systemic involvement (Fig.3).
It is critical
that dental needs be assessed as soon as a definitive diagnosis of leukemia has
been rendered and a decision is made to initiate a radiation, chemotherapy, or
BMT protocol. Unfortunately, oral care has been overlooked in the past, but
aggressive promotion of dental intervention as a part of leukemia treatment
protocols in recent years has dramatically decreased the incidence of oral
complications.
During the acute
phase of the disease only those procedures that are necessary to alleviate the
discomfort and hemorrhaging should be performed. Conversely, during a period of
remission every attempt should be made to achieve a state of periodontal
health. The treatment should be conservative, consisting of the removal of all
local irritants and instruction in good plaque control techniques. The distinct
benefits of strict plaque control in severely granulocytopenic leukemia
patients have been demonstrated: obtaining excellent gingival health and
minimizing oral ulceration throughout chemotherapy.

Fig.4.Ulcerations on lips of a female patient with
graft-versus-host disease (GVHD) after bone marrow transplantation. Severe intraoral
ulcerations also were present on the buccal mucosa, the floor of mouth, and the
ventral surface of the tongue. The patient died within 1 month of this
photograph.
Severe gingival bleeding resulting from thrombocytopenia often can be
managed successfully with localized treatment. The use of an absorbable gelatin
sponge with topical thrombin or placement of microfibrillar collagen is often
sufficient. Some authors report successful management of gingival bleeding with
oral rinses of antifibrinolytic agents. If these measures are not successful in
stopping blood flow from an oral site, platelet transfusions may be necessary.
Management of oral ulcers in patients with leukemia should be directed
toward preventing the spread of localized infection and bacteremia, promoting
healing of the lesion, and decreasing pain. Oral ulcers or extensive tissue
sloughing may serve as the source of life-threatening septicemia in patients
with leukemia (Fig.4). Topical antibacterial and antifungal medication should be used.
Chlorhexidine mouth rinses are effective in reducing the severity of oral
ulcerations, primarily by minimizing secondary infection of these lesions.
Severe ulcers showing clinical signs of infection should be treated with a
combination of topical medication and systemic antibiotics.
Patients with
myelosuppressed leukemia are at risk for a variety of viral infections, most
commonly herpes simplex, varicella zoster, and cytomegalovirus (CMV; Fig.5,6). These infections may become severe and must be
recognized early. Herpes simplex virus and varicella zoster virus respond well
to systemic acyclovir or other antiviral agents, and many patients with
leukemia undergoing chemotherapy are treated prophylactically to prevent
infection.

Fig.5 Herpes zoster. A, Unilateral right palatal ulcerations noted in
68-year-old man. Lesions were acutely painful. Patient had no history of trauma
in this region. B, Ulcerations also noted on labial
mucosa of lower lip. Again, lesions were confined to the patient's
right side. A diagnosis of herpes zoster was made. C, Within 1 week, this patient had major skin ulcerations
on the right side extending across the entire distribution of the trigeminal
nerve. The patient was treated with systemic acyclovir. The lesions resolved
but resulted in significant and prolonged postherpetic neuralgia.

Figure. 6.A 43-year-old male
patient with severe myelosuppression secondary to immunosuppressive drug
therapy. Sloughing of gingiva is apparent around all teeth and affects both
marginal and papillary gingival.
The predominant
inherited coagulation disorders are hemophilia A, hemophilia B, and von Willebrand disease. Coagulopathies may also be
acquired. Liver disease affects coagulation because most of the clotting
factors are synthesized in the liver; thus the clinician should be wary of
coagulation disorders in alcohol abusers and patients with hepatitis. Vitamin K
deficiency, usually associated with long-term antibiotic usage or with
malabsorption syndromes, can result in coagulation problems. Several of the
clotting factors are dependent on vitamin K for their synthesis.
Anemias are
qualitative or quantitative deficiencies of the blood, usually resulting from a
decrease in the number of circulating red blood cells (erythrocytes) or in the
amount of hemoglobin, or from a qualitative change in erythrocytes. The major
categories of anemias include the following:
Normocytic-normochromic
anemia
Macrocytic
hyperchromic anemia
Microcytic
hypochromic anemia
Sickle cell anemia
Aplastic anemia
Aplastic anemia is a form of normocytic-normochromic anemia that
results from a lack of bone marrow production of erythrocytes and other blood
cells. The disorder may be genetic or acquired. The acquired form usually
follows exposure to certain drugs, toxic chemicals, or ionizing radiation. The
severity of the clinical manifestations is directly dependent on the degree of
pancytopenia. Because all bone marrow-derived cells are affected, including
leukocytes and platelets, hemorrhage and infection are the major threats to
patients with aplastic anemia. Oral manifestations include petechiae, gingival swelling and bleeding
(often spontaneous), gingival overgrowth, and herpetic infections. Rapid bone loss has been reported, and
periodontal infections have led to severe, life-threatening systemic infection. Fanconi's anemia is a rare form of aplastic anemia in which
chromosomes break and rearrange easily. Most patients with Fanconi's anemia
have birth defects involving multiple organ systems, and early-onset
periodontitis may be seen.106 BMT may provide the best long-term outcome for individuals with aplastic
anemia.
Pernicious anemia (B-12 deficiency anemia)
Pernicious anemia (B-12 deficiency anemia) (Fig. 7), a form of
macrocytic hyperchromic anemia, is caused by a lack of intrinsic factor, normally
produced by the gastric mucosa. Intrinsic factor is essential to the absorption
of vitamin B12 and to the
formation of erythrocytes. The condition can vary in its clinical severity.
Like many anemias, the complexion may appear pale.Gingival
pallor is also common. The tongue is affected in more than 75% of cases;
atrophy of the papillae leaves the dorsal surface red, shiny, and smooth. It is often painful to eat. Pernicious anemia is treated with vitamin B12 supplementation either orally or by injection.
Fig.7 Pernicious anemia: red and smooth dorsum of the tongue

Fig.8 Plummer–Vinson syndrome: redness and atrophy of
the lingual papillae, associated with angular cheilitis
Iron deficiency anemia
Iron deficiency anemia, a microcytic hypochromic anemia, is the most
common form of anemia. In addition to the presence of hypochromic, microcytic
red blood cells, it is characterized by low iron stores, low serum iron
concentration, and low hemoglobin concentration or hematocrit. Iron deficiency
anemia may result from blood loss, such as an occult gastrointestinal bleed or
excessive menstruation. Oral signs and symptoms are similar to pernicious anemia and primarily
affect the tongue and gingiva. Iron deficiency anemia is present in a disorder
known as Plummer-Vinson syndrome (see above Fig.8) and warrants particular attention. This syndrome is characterized by the
glossitis seen in other forms of iron deficiency anemia, combined with
enlargement of the tongue, ulceration of the oral and esophageal mucosa, and
dysphagia (difficulty swallowing). Patients with
Plummer-Vinson syndrome are at significantly increased risk for esophageal
squamous cell carcinoma and should undergo frequent esophageal endoscopy. Iron supplementation is the key to management of iron deficiency  anemia and may relieve the dysphagia associated
with Plummer-Vinson syndrome.
anemia and may relieve the dysphagia associated
with Plummer-Vinson syndrome.
Sickle cell anemia
Sickle cell anemia is a hereditary hemolytic anemia that is found
almost exclusively in black individuals. An abnormal hemoglobin gene is
present. During conditions of decreased oxygen tension, the red blood cells
change shape and resemble a sickle. This can result in sickle cell crisis, in
which the oxygen-carrying capacity of the erythrocytes is diminished and blood
viscosity is increased. Sickle cell crisis is a life-threatening phenomenon.
Sickle cell anemia may present with pallor of the gingiva and oral mucosa.
Studies have not demonstrated an increased risk for gingivitis or periodontitis
in individuals with sickle cell anemia. However, it is important for the clinician to thoroughly examine the
periodontium of these patients, because acute periodontal infection may
precipitate sickle cell crisis.
Thalassemia
This is a type of
anemia where the haemoglobin of RBC is affected and this is more of racial
disease affecting Italin, Greek, Syrian and American in nature. This is
hereditary disease – a congenital defect of globin synthesis resulting unstable
haemoglobin is formed.

Clinical features:
If it develops in
early stage, it will lead to fatal stage. The child is yellowish with pallor of
skin fever, chills and malaise. Spleenomegaly and hepatomegaly may develop.
Oral manifestations :
An unusual
prominence of the premaxilla and oral mucosa have pale color, maxillary teeth
are irregularly arranged. Intraoral radiography shows peculiar trabecular
pattern of maxilla. Coarsening of trabecula and blurring,
disappearance of other resulting “salt and pepper effect.” Thickening of
diploe of skull. Inner and outer plates become elongated
producing bristals like crew cut or hair on end appearance.
Treatment:
B12, B6 injection with liver extract.
Polycythemia
Abnormal increase
of erythrocytes is called polycythemia. The count reaches to 70,000,000 to
10,000,000 per cu mm .This is accompanied by increased haemoglobin and
hematocrit value.
Clinical features:
Patient will have
headache, tinnitus and visual disturbance. The skin will be reddened. Gastric
complains like gas, pain, belching and peptic ulcer, haemorrhage.
Oral manifestation:
1) Gingiva mucosa,
tongue will be deepened;
2) Gingiva is
congested enlarged and spongy bleeding;
3) Petechia and
ecchymosis are common in mucosa;4) Cyanosis will occur

Fig.9 Polycythemia vera
The disease is
known by many other names, like:
· Cryptogenic polycythemia
· Erythremia
· Erythrocytosis megalosplenica
· Myeloproliferative disorder
· Osler’s disease
· Primary polycythemia
· Polycythemia rubra vera
· Polycythemia with chronic cyanosis – Myelopathic polycythemia
· Splenomegalic polycythemia
· Vaquez’s disease
Treatment:
Over activity of
marrow to produce RBC should be suppressed. Phenyle hydrizane and Myleron are
used to destroy RBC and inhibit production of red blood cell.
Thrombocytopenic
purpura is a blood dyscrasia associated with a decrease in circulating
platelets. Thrombocytopenia of clinical significance exists when the whole
blood platelet count is less than 150,000/mm3, although the precise
limits for normal vary slightly among laboratories. Excessive hemorrhage during or after invasive dental treatment is
often seen with platelet counts less than 50,000/mm3. The most common
manifestation of thrombocytopenic purpura is spontaneous hemorrhage into the
skin and mucous membranes. The disease is also characterized by prolonged
bleeding. Two major forms of thrombocytopenic purpura—primary and
secondary—have been described. Primary
(idiopathic) thrombocytopenic purpura (ITP) (Fig.10) is of unknown
etiology. This is a relatively common form of the disease and may be seen at
any age. Secondary thrombocytopenia is caused by a
known etiologic factor such as chemicals or drugs.
Two forms of ITP
are recognized: acute and chronic. Acute ITP is a self-limited disease that
generally remits permanently without sequel. The onset is usually sudden, with
thrombocytopenia manifested by bruising, bleeding, and petechiae a few days to
several weeks after an otherwise uneventful viral illness. Conversely, chronic ITP
is usually a disease of adults and can be sudden or insidious in onset. It is
more frequent in women than in men, and the course is characterized by
remissions and exacerbations. In both acute and chronic ITP, thrombocytopenia
and its manifestation are the only physical or laboratory abnormalities.
The oral
manifestations of thrombocytopenia may be the first clinical signs of the
disease. Purpura, the most common oral sign, is defined as any escape of blood
into subcutaneous tissues. Purpura includes petechiae, ecchymoses, hemorrhagic
vesicles, and hematomas. These may appear on any mucosal surface and are often
seen on the tongue, lips, and occlusal line of the buccal mucosa secondary to
minor trauma. Purpura may be differentiated from vascular lesions by applying
pressure directly to the area. Because purpura results from blood extravagated
into the tissues, these lesions will not blanch. Other oral signs include
spontaneous gingival hemorrhage and prolonged bleeding after trauma,
toothbrushing, extractions, or periodontal therapy. Similar purpuric findings
are seen on the skin. The patient may have a positive history of epistaxis
(bleeding from the nose), hematuria (blood in urine), melena (darkening of
feces caused by blood pigments), and increased menstrual bleeding.Good oral
hygiene and complete removal of plaque and calculus help to minimize gingival
inflammation and reduce gingival bleeding associated with thrombocytopenia.
Gentle plaque control reduces the risk for bleeding. Periodontal therapy should
be limited unless platelet counts exceed a minimum of 50,000/mm3,
and surgery should be avoided until platelet counts are greater than 80,000/mm3.
Any drug previously associated with the onset of thrombocytopenic episodes
should be avoided. Aspirin and nonsteroidal antiinflammatory agents should also
be avoided, because they may potentiate prolonged bleeding.bleeding, is
generally associated with surgical therapy. On occasion, a patient with a
coagulation disorder will have spontaneous gingival bleeding. This is usually
caused by accumulation of plaque, and its presence emphasizes the importance of
excellent oral hygiene in these patients
Erythremia (Vaquez disease)
Erythremia
or chronic erythromyelosis is characterized with abnormal increasing of red
blood cells-a type of chronic hyperplasia of bone marrow which we called true
red. Those middle and old aged males are often more prone to this disease. The
clinical signs are peculiar plethoric redness of skin and mucosa, especially
neck, cheek, lip, ear, nose and the most at distal part of limbs, hyperemic of
eye conjunctiva, resembles the face of a drunken person. The other symptoms are
such as headache, heaviness in the head, noise in ears, weakness and numbness
of limbs; severe patients may even have impaired vision, narrowing of vision
field, double vision, skin itching, about 1/3 even develop thromboses causing
thrombosis of peripheral, brain and coronary arteries, often caused duodenal
ulcers, rheumatoid arthritis and etc.
The etiology of
this disease is still unknown clearly, considering the increasing of red blood
cells is due to erythrocytosis (increased production of red blood cells or
erythrocytes) and is not due to prolongation of life span of red blood cells. The researches shown that the increasing of red blood cells is
related to the abnormality of hemopoeitic stem cells.
The diagnose is mainly based on clinical symptoms and blood
analysis. The blood volume may increases to 120-240ml/kg (in norm 65-90ml/kg),
hematocrit >50%, slow erythrocyte sedimentation rate, hemoglobin maybe more
than 18-24g/dm, often accompanied by increasing of white blood cells,
erythrocyte network normal or increased, neutrophils and alkaline phosphotase
increased. The regenerative activity of bone marrow is obviously marked
hyperactive and the ratio between neutrophils and young erythrocytes is
decreased. Besides, there are positive result of iron dye and decreased iron
storage in bone marrow.
The modern medical
treatment mainly uses phlebotomy, new cytostatics (marcofan and myelosan) and
radioactive phosphorus (32P).
