CLINICAL PHARMACOLOGY OF NON-STEROID AND OTHER ANTI-INFLAMMATORY AGENTS. CLINICAL PHARMACOLOGY OF STEROIDS
The non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used classes of drugs for the management of acute and chronic pain in dentistry. Their therapeutic efficacy and toxicity are well-documented and provide evidence that NSAIDs generally provide an acceptable therapeutic ratio of pain relief with fewer adverse effects than the opioid-mild analgesic combination drugs that they have largely replaced for most dental applications. The great many studies done with the oral surgery model of acute pain indicate that a single dose of an NSAID is more effective than combinations of aspirin or acetaminophen plus an opioid, with fewer side-effects, thus making it preferable for ambulatory patients. The combination of an NSAID with an opioid generally results in marginal analgesic activity but with an increased incidence of side-effects, which limits its use to patients in whom the NSAID alone results in inadequate analgesia. The selective COX-2 inhibitors hold promise for clinical efficacy with less toxicity from chronic administration and may prove advantageous for the relief of chronic orofacial pain. The use of repeated doses of NSAIDs for chronic orofacial pain should be re-evaluated in light of a lack of documented efficacy and the potential for serious gastrointestinal and renal toxicity with repeated dosing.
Anti-inflammatory effects of NSAIDs
This effect of NSAIDs is due to the inhibition of the enzyme COX, which converts arachidonic acid to prostaglandins, TXA2 and prostacyclin.
Acetylsalicylic acid irreversibly inactivates COX-1 and COX-2 by acetylation of a specific serine residue.
Other NSAIDs reversibly inhibit COX-1 and COX-2
Additional anti-inflammatory mechanism may include:
- Interference with the potentiative action of other mediators of inflammation – bradykinin, histamine, serotonin
- Modulation of T-cell function
- Stabilization of lysosomal membranes
- Inhibition of chemotaxis

Analgesic effect of NSAID s
This effect of NSAIDs is thought to be related to the peripheral inhibition of prostaglandin production, but it may also be due to the inhibition of pain stimuli at a subcortical site.
NDAIDs prevent the potentiating action of prostaglandins on endogenous mediators
of peripheral nerve stimulation ( e.g. bradykinin )
Antipyretic effect of NSAIDs
This effect is believed to be related to inhibition of the interleukin-1 and interleukin-6 induced production of prostaglandins in the hypothalmus and the “resetting” of the termoregulatory system, leading to vasodilation and increased heat loss

Clinical uses of NSAIDs
1) Analgesia
2) Inflammation
3) Antipyresis
4) Antiplatelet effect
5) Cancer preventive agents
Adverse effects of NSAIDs
- Gastrointestinal effects: abdominal pain, gastric and duodenal ulcer, diarrhea, pancreatis
- Gastrointestinal hemorrhage, hepatotoxicity
- Renal effect
– Disturbances of renal function with water and sodium retention
- Inhibition of platelet aggregation
- central symptoms: headache, decreased hearing, tinnitus, dizziness, confusion, depression
- Allergic reactions: asthma, rashes, photosensitivity
Pharmacodynamic interaction NSAIDs with other drugs
- NSAIDs + hypotensive drugs ( β-blockers, ACE-inhhibitors, diuretics ) = ↓ hypotensive effect
- NSAIDs + ehanol = ↑risk of bleeding from gastrointestinal tract
- NSAIDs + ticlopidine or clopidogrel = ↑risk of bleeding
- NSAIDs + lithium = ↑lithium toxicity
- NSAIDs + cylosporine or ACE-inhibitors or takrolimus= ↑nephrotoxicity of drugs
- NSAIDs + fluoroquinolons = ↑ toxic action of fluoroquinolons on CNS
- NSAIDs +oral antidiabetic drugs =↑ risk of hypoglycemia
- NSAIDs + cumarines = ↑risk of bleeding from gastrointestinal tract
Pharmacokinetic interaction NSAIDs with other drugs
- NSAIDs + oral antidiabetic drugs = ↑ risk of hypoglycemia
- NSAIDs + cumarines =↑risk of bleeding
- NSAIDs + corticosteroids = ↑risk gastropathy and bleeding from gastrointestinal tract
- NSAIDs + aminogycosides = ↑ ototoxicity and nephrotoxicity of aminogycosides
- NSAIDs + fenytoine or valproinic acid = ↑action of fenytoine or valproinic acid
- NSAIDs + metotrexat or digoxin = ↑action and ↑ toxicity metotrexat or digoxin
- NSAIDs + tricycles antidepressive drugs neuroleptics or antiarrhytmic drugs or
- selective serotonin reuptake inhibitors ( SSRI ) = ↑ action of drugs
Classification of NSAIDs According to mechanism of action
1) COX-1 selective inhibitors
– Acetylsalicylic acid at low dosage
2) Non selective COX inhibitors
– Acetylsalicylic acid at high dosage
– Diclofenac
– Ibuprofen
– Ketoprofen
– Flurbiprofen
– Indomethacin
– Piroxicam
– Naproxen
3) More COX-2 selective inhibitors
-Nimesulid
– Etodolak
– Meloxicam
– Nabumeton
4) COX-2 selective inhibitors
– Celecoxib
– Etorcoxib
– Valdecoxib
Chemical classification of Non-steroidal anti-inflammatory Drugs (NSAIDs)
A: Salicylates
Acetyl salicylic acid (aspirin), sodium salicylate, Mg salicylate, choline salicylate,
Na thio salicylate, salicyl salicylate
B: Propionic acid derivatives
Ibuprofen, ketoprofen, naproxen, oxaprozin, flurbiprofen
C:Indole acetic acid
Indomethacin, sulindac,
D: Substituted anthranilic acids (Rarely used)
Mefenamic acid, meclofenamate Na
E: Pyrrole alkanoic acid (Rarely used)
Tolmetin
F: Oxicams
Piroxicam, meloxicam
G: Difluorophenyl derivatives
Diflunisal
H: Phenyl acetic acid
Diclofenac
I:Acetic acid derivatives
Etodolac
J: Naphthyl acetic acid prodrugs
Nabumetonre
K: Para-amino phenol derivatives
Acetaminophen
Theraputic classification of NSAID s
A: Analgesics
Aspirin, paracetamol
B: Anti-inflammatory
Indomethacin, naproxen, ibuorofen
C: Anti-coagulants
Aspirin
D: Anti-pyretics
Aspirin, paracetamol, indomethacin, celecoxicv, ibuprofen
E: Inflammatory bowel disease
Sulfasalazine, infiximab
F: Anti-cancer drugs
Methotraxate
G: Anti-malarial
Chloroquine, hydroxychloroquine
H: Tissue transplantation
Cyclosporine
I: Chelating agents in wilson’s disease
Penicillamine
J: Anti-gout drugs
Indomethacin, ibuprofen
W.H.O Classification
A: Drugs with weak anti-inflammatory effect
Acetaminophen
B: Drugs with mild to moderate anti-inflammatory effect
Propionic acid derivatives, anthranilic acid derivatives
C: Drugs with marked anti-inflammatory effects
Salicylates, acetic acid derivatives, oxicams, diclofenac, etodolac
The role of prostaglandins in the pathogenesis of periodontal disease was further confirmed in studies which examined the levels of arachidonic acid metabolites in inflamed gingiva. Using crevicular fluid and samples of gingival tissue, investigators were able to determine that periodontally-diseased tissues had levels of prostaglandins that were significantly elevated compared to levels found in healthy tissues. For example, Goodson et al. noted that when examining levels of prostaglandin E2 in inflamed gingiva, the level was found to be 10-fold higher than that in healthy gingiva. His work was corroborated by El Attar et al. who found PGE2 levels to be increased as much as 20 times in diseased human gingival tissues. Offenbacher found that the crevicular fluid levels of PGE2 were also increased in adult patients with periodontal disease. The other metabolites of the arachidonic pathway, thromboxane, prostacyclin, and the leukotrienes have all been shown by other investigators to be increased in diseased states. A number of prospective studies have been done which confirm the forgoing conclusion. Non-steroidal drugs which were most frequently studied included indomethacin and its analogue Sulindac, the various drugs of the propionic acid group, and piroxicam. Most early work was done with indomethacin, one of the earliest known synthetically-produced, non-steroidal compounds. Indomethacin was used in a wide variety of animal models with ligature-induced periodontitis. Rats, monkeys, dogs, and hamsters are just a few of the animal models used. Nichols and co-workers used indomethacin to treat induced periodontal disease in rats with ligatures. They found that bone metabolism was reduced in those rats treated with indomethacin but not in those untreated. Unfortunately, the reduction did not persist. Using silk ligatures in beagles to produce the disease, Nyman, Schroeder, and Lindhe showed that 1.0 mg/kg indomethacin, when administered daily, slowed the initial inflammatory response as well as osteoclastic bone resorption. Lasfarques and Saffar also induced periodontal disease in golden hamsters. Using 2.0 mg/kg daily indomethacin, they noticed that bone loss was reduced by 28%. Weaks-Dybvig and co-workers induced inflammation by using silk ligatures in squirrel monkeys and administered 5.0 mg/kg indomethacin twice daily. The administration of indomethacin reduced the loss of alveolar bone mass and the loss of bone height. In addition, a decrease in osteoclast density was observed when compared to the untreated controls. This group of studies strongly suggests that indomethacin is able to reduce bone loss due to periodontal disease. It remains unclear what the minimum effective dose would be in these animal systems. Due to the disparity in the method of disease induction as well as animal models used in these studies, it is difficult to make anything other than the most general conclusions from the indomethacin research even though the drug appears to have some promise in reducing the rate of bone loss in periodontal disease. Interestingly, it appears that indomethacin is at its most effective when the animals were plaque-free. As plaque accumulated at the gingival margin, the drug appeared to be less able to decrease the effects of the ongoing inflammatory process suggesting that other factors are important as well.
Ibuprofen, flurbiprofen, and naproxen, are known inhibitors of cyclooxygenase, although there is considerable variation in their potency . Ibuprofen is roughly equivalent to aspirin in potency, while flurbiprofen is more potent than ibuprofen and less potent thaaproxen which is 20 times more potent than aspirin.30 In addition to their inhibition of cyclooxygenase, some of these propionic acid derivatives may also inhibit the migration and other functions of leukocytes.
In a comparison of the effectiveness of flurbiprofen and indomethacin, Williams and co-workers administered systemic flurbiprofen (0.02 mg/kg) and systemic indomethacin (1.0 mg/kg) to adult beagle dogs. The dosages of the two non-steroidal anti-inflammatory drugs were chosen such that over a 24-hour period, their blood levels would be relatively similar. During the treatment period, it was determined that dogs which received flurbiprofen and indomethacin showed decreased rates of bone loss of 62.8% and 23.8% respectively. The rate of decrease in the flurbiprofen group was similar to that seen in the earlier study by Williams and colleagues. Although the observed rate of bone loss was decreased in indomethacin-treated dogs, the rate was not significantly different than that seen in untreated control dogs during the treatment period. The lower rate of effectiveness seen with indomethacin may be due to its lower potency relative to flurbiprofen. Additionally, it was found that the effectiveness of indomethacin in reducing the rate of bone loss increased over time suggesting that indomethacin may take somewhat longer to be effective than flurbiprofen. Perhaps the most interesting observation of this study was made when bone loss activity of individual sites around teeth was assessed. It was found that these drugs are able to keep sites which had been determined to be “inactive” from becoming “active” and that they could also decrease the number of “active” sites. The levels of arachidonic acid metabolites were seen to increase in control animals throughout the study but were reduced in animals treated with either flurbiprofen or indomethacin. This finding further substantiates that these drugs are significant inhibitors of cyclooxygenase metabolites. finally, the Williams group sought to evaluate the effect of topical flurbiprofen on the rate of bone loss compared with the non-steroidal drug ibuprofen, a less potent propionic acid derivative. Flurbiprofen (0.3 mg in 1.0 ml of gel) was formulated in a dosage equivalent to that given systemically in the two previous dog studies. The systemic ibuprofen was administered in a sustained-release preparation at 4.0 mg/kg and in standard formulations at 4.0 mg/kg and 0.4 mg/kg.
Several studies have been set up to investigate the effect of NSAID in the long term treatment of periodontal disease. Jeffrey Ebersole, David Capelli, and Michelle Steffen evaluates the responsiveness of adult periodontitis patients to long term treatment with NSAID flurbiprofen. The results showed that the experimental group demonstrated a subsequent decrease in specific serum antibody when compared to placebo. This response was dosage dependent. Since specific serum antibody correlate to the degree of active periodontal disease, reduction of specific serum antibody can be related to the resolution of periodontal disease. Ray C. Williams et al investigate the effect of flurbiprofen in altering the progression of human alveolar bone loss in the treatment of periodontal disease. In this study, the experimental group was given 50mg of flurbiprofen twice a day for two years after hygienic therapy. Level of alveolar bone loss was identified using sequential radiographs. The results demonstrate that NSAID flurbiprofen cna significantly reduce the rate of alveolar bone loss in humans for a limited period. Four times more alveolar bone was preserved in the group administerec flurbiprofen compared to placebo. M.M Abramsonn investigate the effect of systemic administration of flubiprofen and its withdrawal on crevicular fluid prostaglandin E2 (PGE2), thromboxane B2 (TXB2), and clinical periodontal measurements (plaque index, bleeding index, pocket depth, and attachment level). The test group were given flurbiprofen 300mg/day for a period of 43 days in a 56 day study. Levels of PGE2 and TXB2 were determined by radioimmunoassay. The result showed that flurbiprofen group had decreased PGE2 and TXB2 level in crevicular fluid and a gain of clinical attachment at day 29 and 50 when compared to base line. However, seven days after test subjects discontinued the flurbiprofen admininstration, PGE2 and TXB2 returned to baseline levels, and increase in PAL was no longer present, and an increase in probing depth occurred as compared to base line. Flurbiprofen had no effect on PI, GI, or BI. It is concluded that systemic administration of flubiprofen is a potent inhibitor of human periodontal cyclooxygenase metabolites as measured in crevicular fluid.
Naproxen, another member of the propionic acid derivatives, has received some significant research interest. Naproxen is a very potent NSAID, 20 times more potent than aspirin. The half-life of the drug is approximately 14 hours which makes it more easily administered in humans, requiring fewer dosing periods. In the first assessment of the drug, Johnson et al. found that in examining 102 patients taking either naproxen (500 mg twice a day) or a placebo for a period of 30 days during which they refrained from oral hygiene, neither the naproxen-treated group nor the placebo-treated group were statistically different in the amount of plaque developed, the amount of gingival inflammation, or the amount of gingival bleeding. However, when these patients received a prophylaxis and returned to normal hygiene habits, by day 30 the naproxen-treated group showed enhanced resolution of the gingival inflammation. It was concluded that in the presence of adequate home care by the patient, naproxen could enhance the recovery of inflamed gingival tissues. It is therefore evident that 3 propionic acid derivatives, flurbiprofen, ibuprofen, and naproxen, are all excellent inhibitors of alveolar bone resorption in beagles.
Another compound to receive some recent interest is piroxicam, a leading drug for the treatment of arthritis and related inflammatory disease. In addition to its known effects on prostaglandin synthesis, piroxicam penetrates inflamed tissue easily, has an extended plasma half-life, and minimal toxic side effects. These factors make piroxicam an excellent candidate for local delivery. Howell and co-workers investigated the effect of topical application of piroxicam on developing gingivitis in beagles. During the treatment period, plaque accumulation was uniform among the 3 groups and was not statistically significantly different. However, for the piroxicam-treated dogs both the gingival index and the number of bleeding sites was found to be significantly reduced compared to the control groups throughout the treatment period. The authors concluded that topically-applied piroxicam can significantly inhibit the development of gingival inflammation in beagle dogs.
Another drug has been used to inhibit prostanoid synthesis is Diflunisal. Diflunisal is a long-acting nonacetylated NSAID, belong to the aspirin family (salicylic acid). Contrary to aspirin, it has been shown to effectively block prostanoid biosynthesis with minimal effect on hemostasis. Its half life is 11 hours and with a 1000mg loading dose, its onset of action is comparable to other oral analgesic. Due to the long half life, diflunisal can be administered to the patient twice a day at the dosage of 500mg. James S. Minutello et al. evaluate the effect of preoperative diflunisal for postoperative pain following periodontal surgery. The experimental group was given 1000mg eight hours preoperatively, 500mg of diflunisal one hour preoperatively. The result showed that diflunisal when given preoperatively yield a significant reduction in pain (measure using the Mc Gill Pain Questionaire).
Studies have been conducted to evaluate the effect of NSAIDs compared to the opioid combination. Etodolac, a new NSAID, has been used effectively in dentistry without complication and the side effect as obtained in the opioid combination (respiratory depression, habit forming, nausea, vomiting, constipation, and dizziness). Patricia Tucker, John Smith, and Donald Adams compared the two analgesic regimens (etodolac and acetaminophen with hydrocodone)for the control of psotoperative periodontal discomfort. Patients in the etodolac group received two 300mg capsules 30 minutes prior to surgery and then redosed if needed (prn regimen). Patient who received acetaminophen with hydrocodone was not premedicated and followed the prn regimen. The analgesic efficacy was measured by verbal questionaire. The result showed that both regimen were effective in the treatment of postoperative pain and neither of the regimens had any advantages over the other. However, it is more beneficial to use etodolac for postoperative peridontal surgery discomfort in situations where severe pain is expected over an extended period because of the greater frequency of side effects reported after use of high dose opioids.
In conclusion, NSAIDs have been primarily used to control pain and inflammation causd by inflammatory mediators. They should be used as an adjunctive to mechanical therapy since they also possess several side effects. More control longitudinal studies should be carried out prior to using NSAIDs as the chemical therapy in treatement of periodontal disease.
The cell damage associated with inflammation acts on cell membranes to cause leukocytes to release lysosomal enzymes; arachidonic acid is then liberated from precursor compounds, and various eicosanoids are synthesized. As discussed in, the cyclooxygenase (COX) pathway of arachidonate metabolism produces prostaglandins, which have a variety of effects on blood vessels, on nerve endings, and on cells involved in inflammation. The discovery of cyclooxygenase isoforms (COX-1 and COX-2) led to the concepts that the constitutive COX-1 isoform tends to be homeostatic in function, while COX-2 is induced during inflammation and tends to facilitate the inflammatory response. On this basis, highly selective COX-2 inhibitors have been developed and marketed on the assumption that such selective inhibitors would be safer than nonselective COX-1 inhibitors but without loss of efficacy. The lipoxygenase pathway of arachidonate metabolism yields leukotrienes, which have a powerful chemotactic effect on eosinophils, neutrophils, and macrophages and promote bronchoconstriction and alterations in vascular permeability.
The treatment of patients with inflammation involves two primary goals: first, the relief of pain, which is often the presenting symptom and the major continuing complaint of the patient; and second, the slowing orin theoryarrest of the tissue-damaging process. In rheumatoid arthritis, response to therapy can be quantitated by means of the American College of Rheumatology scoring system values ACR20, ACR50, and ACR70, which denote the percentage of patients showing an improvement of 20%, 50%, or 70% in a global assessment of signs and symptoms.
Reduction of inflammation with nonsteroidal anti-inflammatory drugs (NSAIDs) often results in relief of pain for significant periods. Furthermore, most of the nonopioid analgesics (aspirin, etc) also have anti-inflammatory effects, so they are appropriate for the treatment of both acute and chronic inflammatory conditions.
The glucocorticoids also have powerful anti-inflammatory effects and when first introduced were considered to be the ultimate answer to the treatment of inflammatory arthritis. Although there are increasing data that low-dose corticosteroids have disease-modifying properties, the toxicity associated with chronic corticosteroid therapy usually limits their use except in the control of acute flare-ups of joint disease. The NSAIDs continue to have a significant role in the long-term treatment of arthritis.
Another important group of agents is characterized as disease-modifying antirheumatic drugs (DMARDs). They slow the bone damage associated with rheumatoid arthritis and are thought to affect more basic inflammatory mechanisms than do the NSAIDs. They may also be more toxic than the nonsteroidal anti-inflammatory agents.
The analgesic–antipyretic–anti-inflammatory drug group includes chemically and pharmacologically diverse drugs that share the ability to relieve pain, fever, and/or inflammation, symptoms associated with many injuries and illnesses. Drugs discussed in this chapter include aspirin (acetylsalicylic acid or ASA); the prototype, aspirin-related drugs that are often called nonsteroidal anti-inflammatory drugs (NSAIDs, eg, ibuprofen); acetaminophen; and drugs used to prevent or treat gout and migraine.

Aspirin, NSAIDs, and acetaminophen can also be called antiprostaglandin drugs, because they inhibit the synthesis of prostaglandins. Prostaglandins are chemical mediators found in most body tissues; they help regulate many cell functions and participate in the inflammatory response. They are formed when cellular injury occurs and phospholipids in cell membranes release arachidonic acid. Arachidonic acid is then metabolized by cyclooxygenase enzymes to produce prostaglandins, which act briefly in the area where they are produced and are then inactivated. Prostaglandins exert various and opposing effects in different body tissues (Table 7–1).
Aspirin, NSAIDs, and acetaminophen inactivate cyclooxygenases, the enzymes required for prostaglandin formation (Fig. 7–1). Two forms of cyclooxygenase, called COX-1 and COX-2, have been identified. Aspirin and traditional NSAIDs inhibit both COX-1 and COX-2 enzymes. COX-1 is normally synthesized continuously and present in all tissues and cell types, especially platelets, endothelial cells, the gastrointestinal (GI) tract, and the kidneys. Prostaglandins produced by COX-1 are important in numerous homeostatic functions and are associated with protective effects on the stomach and kidneys. In the stomach, they decrease gastric acid secretion, increase mucus secretion, and regulate blood circulation. In the kidneys, these prostaglandins help to maintain adequate blood flow and function. In the cardiovascular system, the prostaglandins help regulate vascular tone (ie, vasoconstriction and vasodilation) and platelet function. Drug-induced inhibition of these prostaglandins results in the adverse effects associated with aspirin and related drugs, especially gastric irritation, ulceration, and bleeding. Inhibition of COX-1 activity in platelets may be more responsible for GI bleeding than inhibition in gastric mucosa. COX-2 is also normally present in several tissues (eg, brain, bone, kidneys, GI tract, and the female reproductive system).
However, it is thought to occur in small amounts or to be inactive until stimulated by pain and inflammation. In inflamed tissues, COX-2 is induced by inflammatory chemical mediators such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNF alpha). In the GI tract, COX-2 is also induced by trauma and Helicobacter pylori infection, a common cause of peptic ulcer disease. Overall, prostaglandins produced by COX-2 are associated with pain and other signs of inflammation.
Inhibition of COX-2 results in the therapeutic effects of analgesia and anti-inflammatory activity. The COX-2 inhibitor drugs are NSAIDs designed to selectively inhibit COX-2 and relieve pain and inflammation with fewer adverse effects, especially stomach damage. To relieve pain, aspirin acts both centrally and peripherally to block the transmission of pain impulses. Related drugs act peripherally to prevent sensitization of pain receptors to various chemical substances released by damaged cells. To relieve fever, the drugs act on the hypothalamus to decrease its response to pyrogens and reset the “thermostat” at a lower level. For inflammation, the drugs prevent prostaglandins from increasing the pain and edema produced by other substances released by damaged cells. Although these drugs relieve symptoms and contribute greatly to the client’s comfort and quality of life, they do not cure the underlying disorders that cause the symptoms.
Aspirin and traditional NSAIDs also have antiplatelet effects that differ in mechanism and extent. When aspirin is absorbed into the bloodstream, the acetyl portion dissociates, then binds irreversibly to platelet COX-1. This action prevents synthesis of thromboxane A2, a prostaglandin derivative, and thereby inhibits platelet aggregation. A small single dose (325 mg) irreversibly acetylates circulating platelets within a few minutes, and effects last for the lifespan of the platelets (7 to 10 days). Most other NSAIDs bind reversibly with platelet COX-1 so that antiplatelet effects occur only while the drug is present in the blood.
Thus, aspirin has greater effects, but all the drugs except acetaminophen and the COX-2 inhibitors inhibit platelet aggregation, interfere with blood coagulation, and increase the risk of bleeding.
INDICATIONS FOR USE
These drugs are widely used to prevent and treat mild to moderate pain and/or inflammation associated with musculoskeletal disorders (eg, osteoarthritis, tendinitis, gout), headache, dysmenorrhea, minor trauma (eg, athletic injuries such as sprains), minor surgery (eg, dental extraction, episiotomy), and other acute and chronic conditions. Despite many similarities, however, aspirin and other NSAIDs differ in their approved uses. Although aspirin is effective in many disorders, its usage has declined for most indications, largely because of adverse effects on the gastrointestinal tract and the advent of newer drugs. At the same time, lowdose aspirin is increasingly prescribed for clients at risk of myocardial infarction or stroke from thrombosis. This indication stems from its antiplatelet activity and resultant effects on blood coagulation (ie, decreased clot formation).
Some NSAIDs such as ibuprofen (Motrin) and related drugs are widely used as anti-inflammatory agents and analgesics; ketorolac (Toradol), which can be given orally and parenterally, is used only as an analgesic. Most of the other NSAIDs are too toxic to use as analgesics and antipyretics. They are used primarily in rheumatoid arthritis and other musculoskeletal disorders that do not respond to safer drugs. Celecoxib (Celebrex) is also used to treat familial adenomatous polyposis, in which the drug reduces the number of polyps and may decrease risks of colon cancer. Several NSAIDs are formulated as eye drops for use in treating eye disorders.
Acetaminophen, which differs chemically from aspirin and other NSAIDs, is commonly used as an aspirin substitute for pain and fever, but it lacks anti-inflammatory and antiplatelet effects.
CONTRAINDICATIONS TO USE
Contraindications to aspirin and nonselective NSAIDs include peptic ulcer disease, gastrointestinal (GI) or other bleeding disorders, history of hypersensitivity reactions, and impaired renal function. In people who are allergic to aspirin, nonaspirin NSAIDs also are contraindicated because hypersensitivity reactions may occur with any drugs that inhibit prostaglandin synthesis. In children and adolescents, aspirin is contraindicated in the presence of viral infections such as influenza or chickenpox because of its association with Reye’s syndrome.
Selective COX-2 inhibitors are contraindicated for clients with a history of peptic ulcers, GI bleeding, asthma, an allergic reaction to other NSAIDs, or severe renal impairment. In addition, celecoxib and valdecoxib are contraindicated in clients who are allergic to sulfonamides and ketorolac is contraindicated in clients at risk of excessive bleeding. Thus, ketorolac should not be administered during labor and delivery; before or during any major surgery; with suspected or confirmed cerebrovascular bleeding; or to clients who are currently taking aspirin or other NSAIDs.
Over-the-counter (OTC) products containing these drugs are contraindicated for chronic alcohol abusers because of possible liver damage (with acetaminophen) or stomach bleeding (with aspirin, ibuprofen, ketoprofen, or naproxen). The Food and Drug Administration (FDA) requires an alcohol warning on the labels of all OTC pain relievers and fever reducers. This warning states that people who drink three or more alcoholic drinks daily should ask their doctors before taking the products.
Aspirin is the prototype of the analgesic–antipyretic–anti-inflammatory drugs and the most commonly used salicylate. Because it is a nonprescription drug and is widely available, people tend to underestimate its usefulness. It is effective in pain of low to moderate intensity, especially that involving the skin, muscles, joints, and other connective tissue.
It is useful in inflammatory disorders, such as arthritis, but many people prefer drugs that cause less gastric irritation. Regular aspirin tablets are well absorbed after oral administration; their action starts within 15 to 30 minutes, peaks in 1 to 2 hours, and lasts 4 to 6 hours. Taking aspirin with food slows absorption, but also decreases gastric irritation. Absorption of enteric-coated aspirin and rectal suppositories is slower and less complete.
Aspirin is distributed to all body tissues and fluids, including fetal tissues, breast milk, and the central nervous system (CNS). The highest concentrations are found in the plasma, liver, heart, and lungs. In plasma, aspirin binds to albumin (75 to 90%). Aspirin has a short half-life of 15–20 minutes because it is rapidly converted to salicylic acid, an active metabolite. Salicylic acid has a half-life of 2 to 3 hours at low doses and 6 to 12 hours at therapeutic antiinflammatory doses. It undergoes oxidation and conjugation in the liver and its metabolites are excreted through the kidneys. In alkaline urine (eg, pH of 8), renal excretion of salicylate is greatly increased.
Aspirin is a home remedy for headaches, colds, influenza and other respiratory infections, muscular aches, and fever. It can be purchased in plain, chewable, enteric-coated, and effervescent tablets and rectal suppositories. It is not marketed in liquid form because it is unstable in solution.
Diflunisal (Dolobid) is a salicylic acid derivative that differs chemically from aspirin. It is reportedly equal or superior to aspirin in mild to moderate pain, rheumatoid arthritis, and osteoarthritis. Compared with aspirin, it has less antipyretic effect, causes less gastric irritation, and has a longer duration of action.
NSAIDs
Propionic acid derivatives include fenoprofen (Nalfon), flurbiprofen (Ansaid), ibuprofen (Motrin, Advil), ketoprofen (Orudis), naproxen (Naprosyn), and oxaprozin (Daypro). In addition to their use as anti-inflammatory agents, some are used as analgesics and antipyretics. Ibuprofen, ketoprofen, and naproxen are available OTC, with recommended doses smaller and durations of use shorter than those for prescription formulations. Although these drugs are usually better tolerated than aspirin, they are much more expensive and may cause all the adverse effects associated with aspirin and other prostaglandin inhibitors.
Ibuprofen, a commonly used drug, is well absorbed with oral administration. Its action starts in about 30 minutes, peaks in 1 to 2 hours, and lasts 4 to 6 hours. The drug is highly bound (about 99%) to plasma proteins and has a half-life of about 2 hours. It is metabolized in the liver and excreted through the kidneys. It is available by prescription and OTC, in tablets, chewable tablets, capsules, oral suspension, and oral drops, for use by adults and children.
Acetic acid derivatives include indomethacin (Indocin), sulindac (Clinoril), and tolmetin (Tolectin). These drugs have strong anti-inflammatory effects and more severe adverse effects than the propionic acid derivatives. Potentially serious adverse effects include GI ulceration, bone marrow depression, hemolytic anemia, mental confusion, depression, and psychosis. These effects are especially associated with indomethacin; the other drugs were developed in an effort to find equally effective but less toxic derivatives of indomethacin.
Although adverse reactions occur less often with sulindac and tolmetin, they are still common. In addition to other uses, intravenous (IV) indomethacin is approved for treatment of patent ductus arteriosus in premature infants. (The ductus arteriosus joins the pulmonary artery to the aorta in the fetal circulation. When it fails to close, blood is shunted from the aorta to the pulmonary artery, causing severe cardiopulmonary problems.)
Other drugs related to this group are etodolac (Lodine), ketorolac (Toradol), and nabumetone (Relafen). Etodolac reportedly causes less gastric irritation, especially in older adults at high risk for GI bleeding. Ketorolac is used only for pain, and although it can be given orally, its unique characteristic is that it can be given by injection. Parenteral ketorolac reportedly compares with morphine and other opioids in analgesic effectiveness for moderate or severe pain. However, its use is limited to 5 days because it increases the risk of bleeding. Hematomas and wound bleeding have been reported with postoperative use.
Oxicam drugs include meloxicam (Mobic) and piroxicam (Feldene). Meloxicam has a serum half-life of 15 to 20 hours and is excreted about equally through urine and feces. Piroxicam has a half-life of about 50 hours. The long half-lives allow the drugs to be given once daily, but optimal efficacy may not occur for 1 to 2 weeks.
Diclofenac sodium (Voltaren) is chemically different from but pharmacologically similar to other NSAIDs. Formulations are delayed or extended-release and onset of action is therefore delayed. Peak action occurs in about 2 hours and effects last 12 to 15 hours. The formulation of diclofenac potassium (Cataflam) is immediate-release; action starts quickly, peaks in about 20 minutes to 2 hours, and also lasts 12 to 15 hours. As a result, the potassium salt may be given for rapid relief of pain and primary dysmenorrhea. Diclofenac has a serum halflife of about 2 hours and is excreted mainly in the urine.
Cyclooxygenase-2 inhibitors block production of prostaglandins associated with pain and inflammation without blocking those associated with protective effects on gastric mucosa. Thus, they produce less gastric irritation than aspirin and other NSAIDs. In addition, they are not associated with increased risks of bleeding because they do not have the antiplatelet effects of aspirin and other NSAIDs. Despite the relative safety of these drugs, there have been a few cases reported in which hypertension was acutely worsened by the drugs (blood pressure returned to previous levels when the drugs were discontinued; the drugs do not raise blood pressure iormotensive clients) and some clients receiving a COX-2 inhibitor had a small increase in the incidence of myocardial infarction and stroke due to thrombosis, compared with clients receiving a nonselective NSAID (naproxen) or placebo. These concerns are being investigated.
Celecoxib (Celebrex) is well absorbed with oral administration; peak plasma levels and peak action occur approximately 3 hours after an oral dose. It is highly protein bound (97%) and its serum half-life is about 11 hours. It is metabolized by the cytochrome P450 enzymes in the liver to inactive metabolites that are then excreted in the urine. A small amount is excreted unchanged in the urine. Rofecoxib (Vioxx) acts within 45 minutes and peaks in 2 to 3 hours. It is 87% protein bound and has a half-life of 17 hours. It is metabolized in the liver and excreted in urine and feces.
Valdecoxib (Bextra) is a newer COX-2 inhibitor.
Acetaminophen (also called APAP, an abbreviation for N-Acetyl-P-Aminophenol) is a nonprescription drug commonly used as an aspirin substitute because it does not cause nausea, vomiting, or GI bleeding, and it does not interfere with blood clotting. It is equal to aspirin in analgesic and antipyretic effects, but it lacks anti-inflammatory activity. Acetaminophen is well absorbed with oral administration and peak plasma concentrations are reached within 30 to 120 minutes. Duration of action is 3 to 4 hours. Acetaminophen is metabolized in the liver; approximately 94% is excreted in the urine as inactive glucuronate and sulfate conjugates. Approximately 4% is metabolized to a toxic metabolite, which is normally inactivated by conjugation with glutathione and excreted in urine. With usual therapeutic doses, a sufficient amount of glutathione is available in the liver to detoxify acetaminophen. In acute or chronic overdose situations, however, the supply of glutathione may become depleted.
In the absence of glutathione, the toxic metabolite combines with liver cells and causes damage or fatal liver necrosis. In people who abuse alcohol, usual therapeutic doses may cause or increase liver damage. The probable mechanism for increased risk of hepatotoxicity in this population is that ethanol induces drug-metabolizing enzymes in the liver. The resulting rapid metabolism of acetaminophen produces enough toxic metabolite to exceed the available glutathione. Acetaminophen is available in tablet, liquid, and rectal suppository forms and is iumerous combination products marketed as analgesics and cold remedies. It is often prescribed with codeine, hydrocodone, or oxycodone for added analgesic effects.
Introduction
Aspirin’s long use and availability without prescription diminishes its glamour compared with that of the newer NSAIDs. Aspirin is now rarely used as an anti-inflammatory medication; it has been replaced by ibuprofen and naproxen, since they are effective, are also available over the counter, and have good to excellent safety records.
Pharmacokinetics
Salicylic acid is a simple organic acid with a pKa of 3.0. Aspirin (acetylsalicylic acid; ASA) has a pKa of 3.5. Sodium salicylate and aspirin are equally effective anti-inflammatory drugs, though aspirin may be more effective as an analgesic. The salicylates are rapidly absorbed from the stomach and upper small intestine, yielding a peak plasma salicylate level within 1-2 hours. Aspirin is absorbed as such and is rapidly hydrolyzed (serum half-life 15 minutes) to acetic acid and salicylate by esterases in tissue and blood. Salicylate is bound to albumin, but the binding and metabolism of salicylates are saturable so that the unbound fraction increases as total concentration increases. Beyond a total body load of 600 mg, increases in salicylate dosage increase salicylate concentration disproportionately. As doses of aspirin increase, salicylate elimination half-life increases from 3-5 hours (for 600 mg/d dosage) to 12-16 hours (dosage 3.6 g/d). Alkalinization of the urine increases the rate of excretion of free salicylate and its water-soluble conjugates.
A. ANTI-INFLAMMATORY EFFECTS
Aspirin is a nonselective inhibitor of both COX isoforms, but salicylate is much less effective in inhibiting either isoform. Nonacetylated salicylates may work as oxygen radical scavengers. Aspirin irreversibly inhibits COX and inhibits platelet aggregation, while nonacetylated salicylates do not.
Aspirin is most effective in reducing pain of mild to moderate intensity through its effects on inflammation and because it probably inhibits pain stimuli at a subcortical site.
Aspirin’s antipyretic effect is probably mediated by both COX inhibition in the central nervous system and inhibition of interleukin-1 (which is released from macrophages during episodes of inflammation).
Aspirin irreversibly inhibits platelet COX, so that aspirin’s antiplatelet effect lasts 8-10 days (the life of the platelet).
A. ANALGESIA, ANTIPYRESIS, AND ANTI-INFLAMMATORY EFFECTS
Aspirin is employed for mild to moderate pain of varied origin but is not effective for severe visceral pain. Aspirin and other NSAIDs have been combined with opioid analgesics for treatment of cancer pain, where their anti-inflammatory effects act synergistically with the opioids to enhance analgesia. High-dose salicylates are effective for treatment of rheumatic fever, rheumatoid arthritis, and other inflammatory joint conditions.
Aspirin decreases the incidence of transient ischemic attacks, unstable angina, coronary artery thrombosis with myocardial infarction, and thrombosis after coronary artery bypass grafting
Epidemiologic studies suggest that long-term use of aspirin at low dosage is associated with a lower incidence of colon cancer, possibly related to its COX-inhibiting effects.
Dosage
The optimal analgesic or antipyretic dose of aspirin is less than the 0.6-
At the usual dosage, aspirin’s main adverse effects are gastric upset (intolerance) and gastric and duodenal ulcers; hepatotoxicity, asthma, rashes, and renal toxicity occur less frequently. A dose-related increase in fecal blood loss is routinely associated with aspirin administration, although some mucosal adaptation occurs in many patients, so that blood loss declines back to baseline over 4-6 weeks.
With higher doses, patients may experience salicylismvomiting, tinnitus, decreased hearing, and vertigoreversible by reducing the dosage. Still larger doses of salicylates cause hyperpnea through a direct effect on the medulla. At toxic salicylate levels, respiratory alkalosis followed by metabolic acidosis (salicylate accumulation), respiratory depression, and even cardiotoxicity and glucose intolerance can occur. Like other NSAIDs, aspirin can cause elevation of liver enzymes (a frequent but mild effect), hepatitis (rare), decreased renal function, bleeding, rashes, and asthma.
The antiplatelet action of aspirin contraindicates its use by patients with hemophilia. Although previously not recommended during pregnancy, aspirin may be valuable in treating preeclampsia-eclampsia.
Salicylate overdosage constitutes a medical emergency and requires hospitalization
Introduction
These drugs include magnesium choline salicylate, sodium salicylate, and salicylsalicylate. All nonacetylated salicylates are effective anti-inflammatory drugs, although they may be less effective analgesics than aspirin. Because they are much less effective than aspirin as COX inhibitors, they may be preferable when COX inhibition is undesirable, such as in patients with asthma, those with bleeding tendencies, and even (under close supervision) those with renal dysfunction.
The nonacetylated salicylates are administered in the same dosage as aspirin and can be monitored using serum salicylate measurements.
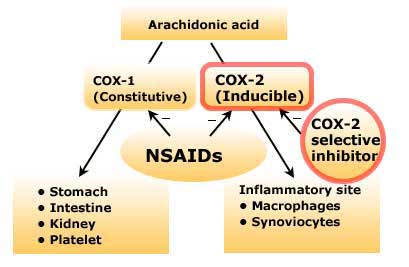
COX-2 selective inhibitors, or coxibs, were developed in an attempt to inhibit prostaglandin synthesis by the COX-2 isoenzyme induced at sites of inflammation without affecting the action of the constitutively active “housekeeping” COX-1 isoenzyme found in the gastrointestinal tract, kidneys, and platelets. Coxibs selectively bind to and block the active site of the COX-2 enzyme much more effectively than that of COX-1. COX-2 inhibitors have analgesic, antipyretic, and anti-inflammatory effects similar to those of nonselective NSAIDs but with an approximate halving of gastrointestinal adverse effects. Likewise, COX-2 inhibitors at usual doses have been shown to have no impact on platelet aggregation, which is mediated by the COX-1 isoenzyme. As a result, COX-2 inhibitors do not offer the cardioprotective effects of traditional nonselective NSAIDs, which has resulted in some patients taking low-dose aspirin in addition to a coxib regimen to maintain this effect. Unfortunately, because COX-2 is constitutively active within the kidney, recommended doses of COX-2 inhibitors cause renal toxicities similar to those associated with traditional NSAIDs. Clinical data have suggested a higher incidence of cardiovascular thrombotic events associated with COX-2 inhibitors such as rofecoxib and valdecoxib, resulting in their withdrawal from the market.
1. Celecoxib
Celecoxib is a selective COX-2 inhibitorabout 10-20 times more selective for COX-2 than for COX-1. Pharmacokinetic and dosage considerations are given in Celecoxib is as effective as other NSAIDs in the treatment of rheumatoid arthritis and osteoarthritis, and in trials it has caused fewer endoscopic ulcers than most other NSAIDs. Probably because it is a sulfonamide, celecoxib may cause rashes. It does not affect platelet aggregation at usual doses. It interacts occasionally with warfarinas would be expected of a drug metabolized via CYP2C9.
Although celecoxib is associated with about half the gastrointestinal side effects of nonselective NSAIDs, the frequency of other adverse effects approximates that of other NSAIDs. Celecoxib causes no more edema or renal effects than other members of the NSAID group, but edema and hypertension have been documented.
2. Etoricoxib
Etoricoxib, a bipyridine derivative, is a second-generation COX-2-selective inhibitor with the highest selectivity ratio of any coxib for inhibition of COX-2 relative to COX-1. It is extensively metabolized by hepatic P450 enzymes followed by renal excretion and has an elimination half-life of 22 hours. Etoricoxib is approved in the United Kingdom for the treatment of the signs and symptoms of osteoarthritis (60 mg once daily) and rheumatoid arthritis (90 mg once daily), for treatment of acute gouty arthritis (120 mg once daily), and for relief of acute musculoskeletal pain (60 mg once daily). Approval in the USA is still pending. Ninety mg daily of etoricoxib has superior efficacy compared with 500 mg of naproxen twice daily in the treatment of rheumatoid arthritis over 12 weeks. Etoricoxib has similar efficacy to traditional NSAIDs for osteoarthritis, acute gouty arthritis, and primary dysmenorrhea and has a gastrointestinal safety profile similar to that of other coxibs. Since etoricoxib has structural similarities to diclofenac, it is appropriate to monitor hepatic function carefully in patients using this drug.
3. Meloxicam
Meloxicam is an enolcarboxamide related to piroxicam that has been shown to preferentially inhibit COX-2 over COX-1, particularly at its lowest therapeutic dose of 7.5 mg/d. It is not as selective as the other coxibs and may be considered “preferentially” selective rather than “highly” selective. The drug is popular in Europe and many other countries for most rheumatic diseases and has recently been approved for treatment of osteoarthritis in the USA. Its efficacy in this condition and rheumatoid arthritis is comparable to that of other NSAIDs. It is associated with fewer clinical gastrointestinal symptoms and complications than piroxicam, diclofenac, and naproxen. Similarly, while meloxicam is known to inhibit synthesis of thromboxane A2, it appears that even at supratherapeutic doses its blockade of thromboxane A2 does not reach levels that result in decreased in vivo platelet function. Other toxicities are similar to those of other NSAIDs.
4. Valdecoxib
Valdecoxib, a diaryl-substituted isoxazole, is a new highly selective COX-2 inhibitor. The analgesic dose for valdecoxib is 20 mg twice daily. Gastrointestinal and other toxicities are similar to those of the other coxibs. Valdecoxib has no effect on platelet aggregation or bleeding time. Serious reactions have been reported in sulfonamide-sensitive individuals. Valdecoxib was withdrawn from the market in the USA in early
1. Diclofenac
Diclofenac is a phenylacetic acid derivative that is relatively nonselective as a COX inhibitor.
Adverse effects occur in approximately 20% of patients and include gastrointestinal distress, occult gastrointestinal bleeding, and gastric ulceration, though ulceration may occur less frequently than with some other NSAIDs. A preparation combining diclofenac and misoprostol decreases upper gastrointestinal ulceration but may result in diarrhea. Another combination of diclofenac and omeprazole was also effective with respect to the prevention of recurrent bleeding, but renal adverse effects were common in high-risk patients. Diclofenac at a dosage of 150 mg/d appears to impair renal blood flow and glomerular filtration rate. Elevation of serum aminotransferases may occur more commonly with this drug than with other NSAIDs.
A 0.1% ophthalmic preparation is recommended for prevention of postoperative ophthalmic inflammation and can be used after intraocular lens implantation and strabismus surgery. A topical gel containing 3% diclofenac is effective for solar keratoses. Diclofenac in rectal suppository form can be considered a drug of choice for preemptive analgesia and postoperative nausea. In Europe, diclofenac is also available as an oral mouthwash and for intramuscular administration.
Although diflunisal is derived from salicylic acid, it is not metabolized to salicylic acid or salicylate. It undergoes an enterohepatic cycle with reabsorption of its glucuronide metabolite followed by cleavage of the glucuronide to again release the active moiety. Diflunisal is subject to capacity-limited metabolism, with serum half-lives at various dosages approximating that of salicylates. In rheumatoid arthritis the recommended dose is 500-1000 mg daily in two divided doses. It is claimed to be particularly effective for cancer pain with bone metastases and for pain control in dental (third molar) surgery. A 2% diflunisal oral ointment is a clinically useful analgesic for painful oral lesions.
Because its clearance depends on renal function as well as hepatic metabolism, diflunisal’s dosage should be limited in patients with significant renal impairment. Its adverse event profile is similar to those of other NSAIDs; pseudoporphyria has also been reported.
3. Etodolac
Etodolac is a racemic acetic acid derivative with an intermediate half-life. It is slightly more COX-2-selective than most other NSAIDs, with a COX-2:COX-1 activity ratio of about 10. Unlike many other racemic NSAIDs, etodolac does not undergo chiral inversion in the body. The dosage of etodolac is 200-400 mg three to four times daily. Etodolac provides good postoperative pain relief after coronary artery bypass operations, although transient impairment of renal function has been reported. There are no data to suggest that etodolac differs significantly from other NSAIDs except in its pharmacokinetic parameters, though it has been claimed to cause less gastric toxicity in terms of ulcer disease than other nonselective NSAIDs.
4. Fenoprofen
Fenoprofen, a propionic acid derivative, is the NSAID most closely associated with interstitial nephritis and is rarely used. Its other toxicities mirror those of other NSAIDs. Fenoprofen is effective for treating the fever, pain, and swelling caused by inflammation. Fenoprofen belongs to a class of drugs called nonsteroidal antiinflammatory drugs (NSAIDs). Other members of the NSAID class of drugs include ibuprofen (Motrin), indomethacin (Indocin), nabumetone (Relafen), naproxen (Aleve) and several others. These drugs are used for the management of mild to moderate pain, fever, and inflammation. They work by reducing the levels of prostaglandins, chemicals that are responsible for the pain, fever, and swelling of inflammation. Fenoprofen blocks the enzymes that make prostaglandins (cyclooxygenases), resulting in lower concentrations of prostaglandins. As a consequence, inflammation, swelling, pain and fever are reduced. Fenoprofen was approved by the FDA in March 1976.
PREPARATIONS: Capsule: 200 mg; Tablet: 600 mg
PRESCRIBED FOR: Fenoprofen is used for the treatment of inflammation and pain due to rheumatoid arthritis and osteoarthritis. Fenoprofen also is used for the relief of mild to moderate pain, such as menstrual cramps, tendinitis, and bursitis.
DOSING: The recommended adult dose for mild to moderate pain is 200 mg every 4-6 hours. Rheumatoid arthritis and osteoarthritis are treated with 600 mg 3-4 times daily. The maximum daily dose is 3200 mg daily. Fenoprofen should be administered with meals in order to avoid stomach upset.
DRUG INTERACTIONS: Fenoprofen is associated with several suspected or probable interactions that affect the action of other drugs. The following examples are the most commonly suspected interactions.
Fenoprofen may increase the blood levels of lithium (Eskalith, Lithobid) by reducing the excretion of lithium by the kidneys. Increased levels of lithium may lead to lithium toxicity.
Fenoprofen may reduce the blood pressure lowering effects of blood pressure medications. This may occur because prostaglandins play a role in the regulation of blood pressure.
When NSAIDs are used in combination with methotrexate (Rheumatrex, Trexall) or aminoglycosides (for example, gentamicin) the blood levels of the methotrexate or aminoglycoside may increase, presumably because their elimination from the body is reduced. This may lead to more methotrexate or aminoglycoside-related side effects.
Individuals taking oral blood thinners or anticoagulants, for example, warfarin, (Coumadin), should avoid fenoprofen because fenoprofen also thins the blood, and excessive blood thinning may lead to bleeding.
Persons who have more than three alcoholic beverages per day are at increased risk of developing stomach ulcers when taking fenoprofen or other NSAIDs.
PREGNANCY: There are no adequate studies of fenoprofen in pregnant women. Therefore, fenoprofen is not recommended during pregnancy.
NURSING MOTHERS: It is not known if fenoprofen is excreted in breast milk.
SIDE EFFECTS: Common side effects include rash, ringing in the ears, headaches, dizziness, drowsiness, abdominal pain, nausea, diarrhea, constipation, heartburn, fluid retention and dry mouth.
NSAIDs reduce the ability of blood to clot and therefore increase bleeding after an injury. Fenoprofen also may cause stomach and intestinal bleeding and ulcers. Sometimes, stomach ulceration and intestinal bleeding may occur without any abdominal pain. Black tarry stools, weakness, and dizziness upon standing (orthostatic hypotension) may be the only signs of the bleeding.
People who are allergic to other NSAIDs should not use fenoprofen. NSAIDs reduce the flow of blood to the kidneys and impair function of the kidneys. The impairment is most likely to occur in patients with preexisting impairment of kidney function or congestive heart failure, and use of NSAIDs in these patients should be done cautiously. Individuals with asthma are more likely to experience allergic reactions to fenoprofen and other NSAIDs. Fluid retention, blood clots, heart attacks, hypertension, and heart failure also have been associated with the use of NSAIDs.
5. Flurbiprofen
Flurbiprofen is a propionic acid derivative with a possibly more complex mechanism of action than other NSAIDs. Its (S)(-) enantiomer inhibits COX nonselectively, but it has been shown in rat tissue to also affect TNF- and nitric oxide synthesis. Hepatic metabolism is extensive; its (R)(+) and (S)(-) enantiomers are metabolized differently, and it does not undergo chiral conversion. It does demonstrate enterohepatic circulation.
The efficacy of flurbiprofen at dosages of 200-400 mg/d is comparable to that of aspirin and other NSAIDs in clinical trials for patients with rheumatoid arthritis, ankylosing spondylitis, gout, and osteoarthritis. It is also available in a topical ophthalmic formulation for inhibition of intraoperative miosis. Flurbiprofen intravenously has been found to be effective for perioperative analgesia in minor ear, neck, and nose surgery and in lozenge form for sore throat.
Although its adverse effect profile is similar to that of other NSAIDs in most ways, flurbiprofen is also associated rarely with cogwheel rigidity, ataxia, tremor, and myoclonus.
6. Ibuprofen
Ibuprofen is a simple derivative of phenylpropionic acid. In doses of about 2400 mg daily, ibuprofen is equivalent to
Gastrointestinal irritation and bleeding occur, although less frequently than with aspirin. The use of ibuprofen concomitantly with aspirin may decrease the total anti-inflammatory effect. The drug is relatively contraindicated in individuals with nasal polyps, angioedema, and bronchospastic reactivity to aspirin. In addition to the gastrointestinal symptoms (which can be modified by ingestion with meals), rash, pruritus, tinnitus, dizziness, headache, aseptic meningitis (particularly in patients with systemic lupus erythematosus), and fluid retention have been reported. Interaction with anticoagulants is uncommon.
The concomitant administration of ibuprofen antagonizes the irreversible platelet inhibition induced by aspirin. Thus, treatment with ibuprofen in patients with increased cardiovascular risk may limit the cardioprotective effects of aspirin. Rare hematologic effects include agranulocytosis and aplastic anemia. Effects on the kidney (as with all NSAIDs) include acute renal failure, interstitial nephritis, and nephrotic syndrome, but these occur very rarely. Finally, hepatitis has been reported.
7. Indomethacin
Indomethacin, introduced in 1963, is an indole derivative. It is a potent nonselective COX inhibitor and may also inhibit phospholipase A and C, reduce neutrophil migration, and decrease T cell and B cell proliferation. Probenecid prolongs indomethacin’s half-life by inhibiting both renal and biliary clearance. It differs somewhat from other NSAIDs in its indications and toxicities.
Indomethacin is indicated for use in rheumatic conditions and is particularly popular for gout and ankylosing spondylitis. In addition, it has been used to treat patent ductus arteriosus. Indomethacin has been tried in numerous small or uncontrolled trials for many other conditions, including Sweet’s syndrome, juvenile rheumatoid arthritis, pleurisy, nephrotic syndrome, diabetes insipidus, urticarial vasculitis, postepisiotomy pain, and prophylaxis of heterotopic ossification in arthroplasty. An ophthalmic preparation seems to be efficacious for conjunctival inflammation and to reduce pain after traumatic corneal abrasion. Gingival inflammation is reduced after administration of indomethacin oral rinse. Epidural injections produce a degree of pain relief similar to that achieved with methylprednisolone in postlaminectomy syndrome.
At higher dosages, at least a third of patients have reactions to indomethacin requiring discontinuance. The gastrointestinal effects may include abdominal pain, diarrhea, gastrointestinal hemorrhage, and pancreatitis. Headache is experienced by 15-25% of patients and may be associated with dizziness, confusion, and depression. Rarely, psychosis with hallucinations has been reported. Hepatic abnormalities are rare. Serious hematologic reactions have beeoted, including thrombocytopenia and aplastic anemia. Hyperkalemia has been reported and is related to inhibition of the synthesis of prostaglandins in the kidney. Renal papillary necrosis has also been observed. A number of interactions with other drugs have been reported.
8. Ketoprofen
Ketoprofen is a propionic acid derivative that inhibits both COX (nonselectively) and lipoxygenase. Concurrent administration of probenecid elevates ketoprofen levels and prolongs its plasma half-life.
The effectiveness of ketoprofen at dosages of 100-300 mg/d is equivalent to that of other NSAIDs in the treatment of rheumatoid arthritis, osteoarthritis, gout, dysmenorrhea, and other painful conditions. In spite of its dual effect on prostaglandins and leukotrienes, ketoprofen is not superior to other NSAIDs. Its major adverse effects are on the gastrointestinal tract and the central nervous system.
Ketorolac is an NSAID promoted for systemic use mainly as an analgesic, not as an anti-inflammatory drug (although it has typical NSAID properties). The drug is an effective analgesic and has been used successfully to replace morphine in some situations involving mild to moderate postsurgical pain. It is most often given intramuscularly or intravenously, but an oral dose formulation is available. When used with an opioid, it may decrease the opioid requirement by 25-50%. An ophthalmic preparation is available for ocular inflammatory conditions. Toxicities are similar to those of other NSAIDs, although renal toxicity may be more common with chronic use.
10. Meclofenamate Mefenamic Acid
Meclofenamate and mefenamic acid inhibit both COX and phospholipase A2. They are rarely used today.
Side Effects agranulocytosis, anemia, aplastic anemia, azotemia, bullous rash, diarrhea, dizziness, elevated hepatic enzymes, GI bleeding, GI perforation, headache, heart failure, hematuria, hemolysis, interstitial nephritis, jaundice, maculopapular rash, nephrotic syndrome, pancytopenia, peripheral edema, platelet dysfunction, proteinuria, renal papillary necrosis, toxic epidermal necrolysis, urticaria, Drug-Vitamin-Herb InteractionsNegative interactions: Arginine Arginine stimulates gastrin production which increases stomach acid. This can irritate the stomach.
Feverfew Feverfew has similar actions. Combining Feverfew with NSAIDs may increase the risk of side effects. Natural blood thinners: Garlic, Ginkgo, Vitamin E, Policosanol, Dong quai, Ginger, Horse chestnut, Red clover, Fish oils, and OPCs. Natural blood thinners may increase the tendency to bleed, a side effect of NSAIDs. Potassium, magnesium, and calcium citrate Citrates alkalinize the urine which decrease the therapeutic effect of NSAIDs and ASA. St JohnÕs Wort and Dong quai St JohnÕs Wort and Dong Quai can cause sensitivity to sunlight, which may add to side effects of NSAIDs. Positive interactions: Cayenne Cayenne may help prevent gastric bleeding. Colostrum Colostrum may help protect against ulcers caused by NSAIDs. Folate Folate may be depleted by NSAIDs. Licorice (DGL) Licorice (DGL) may help protect the stomach against gastric damage caused by NSAIDs. Vitamin C Vitamin C may be depleted by NSAIDs.
11. Nabumetone
Nabumetone is the only nonacid NSAID in current use; it is converted to the active acetic acid derivative in the body. It is given as a ketone prodrug that resembles naproxen in structure. Its half-life of more than 24 hours permits once-daily dosing, and the drug does not appear to undergo enterohepatic circulation. Renal impairment results in a doubling of its half-life and a 30% increase in the area under the curve. Its properties are very similar to those of other NSAIDs, though it may be less damaging to the stomach than some other NSAIDs when given at a dosage of 1000 mg/d. Unfortunately, higher doses (eg, 1500-2000 mg/d) are ofteeeded, and this is a very expensive NSAID. Like naproxen, nabumetone has been reported to cause pseudoporphyria and photosensitivity in some patients. Other adverse effects mirror those of other NSAIDs.
12. Naproxen
Naproxen is a naphthylpropionic acid derivative. It is the only NSAID presently marketed as a single enantiomer, and it is a nonselective COX inhibitor. Naproxen’s free fraction is significantly higher in women than in men, although albumin binding is very high in both sexes. Naproxen is effective for the usual rheumatologic indications and is available both in a slow-release formulation and as an oral suspension. A topical preparation and an ophthalmic solution are also available.The incidence of upper gastrointestinal bleeding in over-the-counter use is low but still double that of over-the-counter ibuprofen (perhaps due to a dose effect). Rare cases of allergic pneumonitis, leukocytoclastic vasculitis, and pseudoporphyria as well as the more common NSAID-associated adverse effects have beeoted.
13. Oxaprozin
Oxaprozin is another propionic acid derivative NSAID. Its major difference from the other members of this subgroup is a very long half-life (50-60 hours), although oxaprozin does not undergo enterohepatic circulation. The drug has the same benefits and risks that are associated with other NSAIDs. It is mildly uricosuric, making it potentially more useful in gout than some other NSAIDs.
DAYPRO ALTA (oxaprozin potassium tablets) is a member of the propionic acid group of nonsteroidal anti-inflammatory drugs (NSAIDs). Each blue, capsule-shaped tablet contains oxaprozin potassium (678mg equivalent to 600mg of oxaprozin) for oral administration. The chemical name for oxaprozin potassium is 4,5-diphenyl-2-oxazolepropionic acid, potassium salt. Its empirical formula is C18H14NO3K and molecular weight is 331. Oxaprozin potassium is a white to off white powder with a melting point of
It has the following structural formula:
|
|
Inactive ingredients in DAYPRO ALTA (oxaprozin) tablets include microcrystalline cellulose, hydroxypropyl methylcellulose, pregelatinized corn starch, stearic acid, colloidal silicon dioxide, polyethylene glycol, titanium dioxide, FD&C Blue #1 Aluminum Lake, and pharmaceutical glaze
Side effects
Get emergency medical help if you have any of these signs of an allergic reaction: hives; difficulty breathing; swelling of your face, lips, tongue, or throat.
Stop taking oxaprozin and seek medical attention or call your doctor at once if you have any of these serious side effects:
- chest pain, weakness, shortness of breath, slurred speech, problems with vision or balance;
- black, bloody, or tarry stools, coughing up blood or vomit that looks like coffee grounds;
- urinating less than usual or not at all;
- nausea, stomach pain, low fever, loss of…
14. Phenylbutazone
DESCRIPTION: Phenylbutazone 20% Injection (Phenylbutazone) is a synthetic, nonhormonal anti-inflammoatory, antipyretic compound useful in the management of inflammaotry conditions. The apparent analgesic effect is probably related mainly to the compound’s anti-inflammatory properties. Chemically, Phenylbutazone is 4-butyl-1,2-diphenyl-3,5-pyrazolidinedione. It is a pyrazolon derivative entirely unrelated to the steroid hormones, and has the following structural formula:
INDICATIONS
For relief of inflammatory conditions associated with the musculockeletal system in horses.
Contraindications
Treated animals should not be slaughtered for food purposes. Parenteral injections should be made intravenously only; do not inject subcutaneously or intramuscularly. Use with caution in patients who have a history of drug allergy.
PRECAUTIONS
Stop medication at the first sign of gastrointestinal upset, jaundice, or blood dyscrasia. Authenticated cases of agranulocytosis associated with the drug have occurred in man. To guard against this possibility, conduct routine blood counts at weekly intervals of two weeks thereafter. Any significant fall in th total white count, relative decrease in granulocytes, or black or tarry stools, should be regarded as a signal for immediate cessation of therapy and institution of appropriate counter measures. In the treatment of inflammatory conditions associated with infections, specific anti-infective therapy is required.
Store in refrigerator between 2o – 8oC (36o – 46oF)
Phenylbutazone Dosage and Administration
INTRAVENOUSLY: 1 to
GUIDELINES TO SUCCESSFUL THERAPY 1. Use a relatively high dose for the first 48 hours, then reduce gradually to a maintenance dose. Maintain lowest dose capable of producing desired clinical response.
2. Response to Phenylbutazone therapy is prompt, usually occurring within 24 hours. If no significant clinical response is evident after 5 days, reevaluate diagnosis and therapeutic approach.
3. In animals, Phenylbutazone is largely metabolized in 8 hours. It is recommended that a third of the daily dose be administered at 8 hour intervals. Reduce dosage as symptoms regress. In some cases, treatment may be given only when symptoms appear with no need for continuous medication. If long-term therapy is planned, oral administration is suggested.
4. Many chronic conditions will respond to Phenylbutazone therapy, but discontinuance of treatment may result in recurrence of symptoms.
15. Piroxicam
Piroxicam, an oxicam, is a nonselective COX inhibitor that at high concentrations also inhibits polymorphonuclear leukocyte migration, decreases oxygen radical production, and inhibits lymphocyte function. Its long half-life permits once-daily dosing.
Piroxicam can be used for the usual rheumatic indications. Toxicity includes gastrointestinal symptoms (20% of patients), dizziness, tinnitus, headache, and rash. When piroxicam is used in dosages higher than 20 mg/d, an increased incidence of peptic ulcer and bleeding is encountered. Epidemiologic studies suggest that this risk is as much as 9.5 times higher with piroxicam than with other NSAIDs.
16. Sulindac
Sulindac is a sulfoxide prodrug. It is reversibly metabolized to the active sulfide metabolite, which is excreted in bile and then reabsorbed from the intestine. The enterohepatic cycling prolongs the duration of action to 12-16 hours. The indications and adverse reactions of sulindac are similar to those of other NSAIDs. In addition to its rheumatic disease indications, sulindac suppresses familial intestinal polyposis; it may inhibit the development of colon, breast, and prostate cancer in humans. It appears to inhibit the occurrence of gastrointestinal cancer in rats. The latter effect may be caused by the sulfone rather than the sulfide.
Among the more severe adverse reactions, Stevens-Johnson epidermal necrolysis syndrome, thrombocytopenia, agranulocytosis, and nephrotic syndrome have all been observed. Like diclofenac, sulindac may have some propensity to cause elevation of serum aminotransferases; it is also sometimes associated with cholestatic liver damage, which disappears or becomes quiescent when the drug is stopped.
Side effects
Get emergency medical help if you have any of these signs of an allergic reaction: hives; difficulty breathing; swelling of your face, lips, tongue, or throat.
Stop using sulindac and call your doctor at once if you have a serious side effect such as:
- chest pain, weakness, shortness of breath, slurred speech, problems with vision or balance;
- black, bloody, or tarry stools, coughing up blood or vomit that looks like coffee grounds;
- urinating less than usual or not at all;
- nausea, stomach pain, low fever, loss of appetite, dark urine, clay-colored…
Precautions
Before taking sulindac, tell your doctor or pharmacist if you are allergic to it; or to aspirin or other NSAIDs (such as ibuprofen, naproxen, celecoxib); or if you have any other allergies. This product may contain inactive ingredients, which can cause allergic reactions or other problems. Talk to your pharmacist for more details.
Before using this medication, tell your doctor or pharmacist your medical history, especially of: asthma (including a history of worsening breathing after taking aspirin or other NSAIDs), bleeding or clotting problems, dehydration, growths in the nose (nasal polyps), heart disease (such as congestive heart failure, previous heart attack), high blood pressure, kidney disease, kidney stones, liver disease,..etc).
17. Tenoxicam
Tenoxicam is an oxicam similar to piroxicam and shares its nonselective COX inhibition, long half-life (72 hours), efficacy, and toxicity profile. It is available abroad but not in the USA.
Tiaprofen is a racemic propionic acid derivative but does not undergo stereoconversion. It has a short serum half-life (1-2 hours) with an increase to 2-4 hours in the elderly. This drug inhibits renal uric acid reabsorption and thus decreases serum uric acid slightly. It is available for oral and intramuscular administration. Its efficacy and adverse event profiles mirror those of other NSAIDs. Tiaprofen is not available in the USA.
19. Tolmetin
Tolmetin is a nonselective COX inhibitor with a short half-life (1-2 hours)and is not often used. Its efficacy and toxicity profiles are similar to those of other NSAIDs with the following exceptions: it is ineffective (for unknown reasons) in the treatment of gout, and it may cause (rarely) thrombocytopenic purpura.
20. Azapropazone Carprofen
These drugs are available in many other countries but are not sold in the USA. Azapropazone (apazone), a pyrazolone derivative, is structurally related to phenylbutazone but appears less likely to cause agranulocytosis. Its half-life of 12-16 hours may be doubled in patients with decreased renal function. Carprofen is a propionic acid derivative with a half-life of 10-16 hours. The indications and adverse effects of azapropazone and carprofen are similar to those of other NSAIDs.
CLINICAL PHARMACOLOGY OF THE NSAIDS
All NSAIDs, including aspirin, are about equally efficacious with a few exceptionstolmetin seems not to be effective for gout, and aspirin is less effective than other NSAIDs (eg, indomethacin) for ankylosing spondylitis. Thus, NSAIDs tend to be differentiated on the basis of toxicity and cost-effectiveness. For example, the gastrointestinal and renal side effects of ketorolac limit its use. Some surveys suggest that indomethacin, tolmetin, and meclofenamate are the NSAIDs associated with the greatest toxicity, while salsalate, aspirin, and ibuprofen are least toxic. The selective COX-2 inhibitors were not included in this analysis.
For patients with renal insufficiency, nonacetylated salicylates may be best. Fenoprofen is less often used because of its rare association with interstitial nephritis. Diclofenac and sulindac are associated with more liver function test abnormalities than other NSAIDs. The relatively expensive, selective COX-2 inhibitors are probably safest for patients at high risk for gastrointestinal bleeding but may have a higher risk of cardiovascular toxicity. Celecoxib or a nonselective NSAID plus omeprazole or misoprostol may be appropriate in patients at highest risk for gastrointestinal bleeding; in this subpopulation of patients, they are cost-effective despite their high acquisition costs.
The choice of an NSAID thus requires a balance of efficacy, cost-effectiveness, safety, and numerous personal factors (eg, other drugs also being used, concurrent illness, compliance, medical insurance coverage), so that there is no best NSAID for all patients. There may, however, be one or two best NSAIDs for a specific person.
GLUCOCORTICOIDS
The term corticosteroids actually means all secretions of the adrenal cortex, but it is most often used to designate the glucocorticoids. Glucocorticoids include cortisol, corticosterone, and cortisone. Cortisol accounts for at least 95% of glucocorticoid activity, and approximately 15 to 20 mg are secreted daily. Corticosterone has a small amount of activity, and approximately 1.5 to 4 mg are secreted daily. Cortisone has little activity and is secreted in minute quantities. Glucocorticoids are secreted cyclically, with the largest amount being produced in the early morning and the smallest amount during the evening hours (in people with a normal day–night schedule). At the cellular level, glucocorticoids account for most of the characteristics and physiologic effects of the corticosteroids.
Most of the known effects of the glucocorticoids are mediated by widely distributed glucocorticoid receptors. These proteins are members of the superfamily of nuclear receptors that includes steroid, sterol (vitamin D), thyroid, retinoic acid, and many other receptors with unknown or nonexistent ligands (orphan receptors). All these receptors interact with the promoters ofand regulate the transcription oftarget genes. In the absence of the hormonal ligand, glucocorticoid receptors are primarily cytoplasmic, in oligomeric complexes with heat shock proteins (Hsp). The most important of these are two molecules of Hsp90, although other proteins are certainly involved. Free hormone from the plasma and interstitial fluid enters the cell and binds to the receptor, inducing conformational changes that allow it to dissociate from the heat shock proteins. The ligand-bound receptor complex then is actively transported into the nucleus, where it interacts with DNA and nuclear proteins. As a homodimer, it binds to glucocorticoid receptor elements (GRE) in the promoters of responsive genes. The GRE is composed of two palindromic sequences that bind to the hormone receptor dimer.
In addition to binding to GREs, the ligand-bound receptor also forms complexes with and influences the function of other transcription factors, such as AP1 and NF-B, which act oon-GRE-containing promoters, to contribute to the regulation of transcription of their responsive genes. These transcription factors have broad actions on the regulation of growth factors, proinflammatory cytokines, etc, and to a great extent mediate the anti-growth, anti-inflammatory, and immunosuppressive effects of glucocorticoids.
Two genes for the corticoid receptor have been identified, one encoding the classic glucocorticoid receptor and the other the mineralocorticoid receptor. Alternative splicing of human glucocorticoid receptor pre-mRNA generates two highly homologous isoforms, termed hGR alpha and hGR beta. hGR alpha is the classic ligand-activated glucocorticoid receptor which, in the hormone-bound state, modulates the expression of glucocorticoid-responsive genes. In contrast, hGR beta does not bind glucocorticoids and is transcriptionally inactive. However, hGR beta is able to inhibit the effects of hormone-activated hGR alpha on glucocorticoid-responsive genes, playing the role of a physiologically relevant endogenous inhibitor of glucocorticoid action.
The glucocorticoid receptor is composed of about 800 amino acids and can be divided into three functional domains. The glucocorticoid-binding domain is located at the carboxyl terminal of the molecule and is the area where free glucocorticoids bind. The DNA-binding domain is located in the middle of the protein and contains nine cysteine residues. This region folds into a “two-finger” structure stabilized by zinc ions connected to cysteines to form two tetrahedrons. This part of the molecule binds to the GREs that regulate glucocorticoid action on glucocorticoid-regulated genes. The zinc-fingers represent the basic structure by which the DNA-binding domain recognizes specific nucleic acid sequences. The amino-terminal domain is involved in the transactivation activity of the receptor and increases its specificity.
The interaction of glucocorticoid receptors with GREs or other transcription factors is facilitated or inhibited by several families of proteins called steroid receptor coregulators, divided into coactivators and corepressors. The coregulators do this by serving as bridges between the receptors and other nuclear proteins and by expressing enzymatic activities such as histone acetylase or deacetylase that alter the conformation of nucleosomes and the transcribability of genes.
Between 10% and 20% of expressed genes in a cell are regulated by glucocorticoids. The number and affinity of receptors for the hormone, the complement of transcription factors and coregulators, and posttranscription events determine the relative specificity of these hormones’ actions in various cells. The effects of glucocorticoids are mainly due to proteins synthesized from mRNA transcribed by their target genes.
Some of the effects of glucocorticoids can be attributed to their binding to aldosterone receptors (ARs). Indeed, ARs bind aldosterone and cortisol with similar affinity. A mineralocorticoid effect of cortisol is avoided in some tissues by expression of 11-hydroxysteroid dehydrogenase type 2, the enzyme responsible for biotransformation to its 11-keto derivative (cortisone), which has minimal affinity for aldosterone receptors.
Prompt effects such as initial feedback suppression of pituitary ACTH occur in minutes and are too rapid to be explained on the basis of gene transcription and protein synthesis. It is not known how these effects are mediated. Among the proposed mechanisms are direct effects on cell membrane receptors for the hormone or nongenomic effects of the classic hormone-bound glucocorticoid receptor. The putative membrane receptors might be entirely different from the known intracellular receptors.
The glucocorticoids have widespread effects because they influence the function of most cells in the body. The major metabolic consequences of glucocorticoid secretion or administration are due to direct actions of these hormones in the cell. However, some important effects are the result of homeostatic responses by insulin and glucagon. Although many of the effects of glucocorticoids are dose-related and become magnified when large amounts are administered for therapeutic purposes, there are also other effectscalled permissive effectsin the absence of which many normal functions become deficient. For example, the response of vascular and bronchial smooth muscle to cate-cholamines is diminished in the absence of cortisol and restored by physiologic amounts of this glucocorticoid. Furthermore, the lipolytic responses of fat cells to catecholamines, ACTH, and growth hormone are attenuated in the absence of glucocorticoids.
The glucocorticoids have important dose-related effects on carbohydrate, protein, and fat metabolism. The same effects are responsible for some of the serious adverse effects associated with their use in therapeutic doses. Glucocorticoids stimulate and are required for gluconeogenesis and glycogen synthesis in the fasting state. They stimulate phosphoenolpyruvate carboxykinase, glucose-6-phosphatase, and glycogen synthase and the release of amino acids in the course of muscle catabolism.
Glucocorticoids increase serum glucose levels and thus stimulate insulin release and inhibit the uptake of glucose by muscle cells, while they stimulate hormone-sensitive lipase and thus lipolysis. The increased insulin secretion stimulates lipogenesis and to a lesser degree inhibits lipolysis, leading to a net increase in fat deposition combined with increased release of fatty acids and glycerol into the circulation.
The net results of these actions are most apparent in the fasting state, when the supply of glucose from gluconeogenesis, the release of amino acids from muscle catabolism, the inhibition of peripheral glucose uptake, and the stimulation of lipolysis all contribute to maintenance of an adequate glucose supply to the brain.
D. CATABOLIC AND ANTIANABOLIC EFFECTS
Although glucocorticoids stimulate RNA and protein synthesis in the liver, they have catabolic and antianabolic effects in lymphoid and connective tissue, muscle, peripheral fat, and skin. Supraphysiologic amounts of glucocorticoids lead to decreased muscle mass and weakness and thinning of the skin. Catabolic and antianabolic effects on bone are the cause of osteoporosis in Cushing’s syndrome and impose a major limitation in the long-term therapeutic use of glucocorticoids. In children, glucocorticoids reduce growth. This effect may be partially prevented by administration of growth hormone in high doses.
E. ANTI-INFLAMMATORY AND IMMUNOSUPPRESSIVE EFFECTS
Glucocorticoids dramatically reduce the manifestations of inflammation. This is due to their profound effects on the concentration, distribution, and function of peripheral leukocytes and to their suppressive effects on the inflammatory cytokines and chemokines and on other mediators of inflammation. Inflammation, regardless of its cause, is characterized by the extravasation and infiltration of leukocytes into the affected tissue. These events are mediated by a complex series of interactions of white cell adhesion molecules with those on endothelial cells and are inhibited by glucocorticoids. After a single dose of a short-acting glucocorticoid, the concentration of neutrophils in the circulation increases while the lymphocytes (T and B cells), monocytes, eosinophils, and basophils decrease. The changes are maximal at 6 hours and are dissipated in 24 hours. The increase ieutrophils is due both to the increased influx into the blood from the bone marrow and decreased migration from the blood vessels, leading to a reduction in the number of cells at the site of inflammation. The reduction in circulating lymphocytes, monocytes, eosinophils, and basophils is primarily the result of their movement from the vascular bed to lymphoid tissue.
Glucocorticoids also inhibit the functions of tissue macrophages and other antigen-presenting cells. The ability of these cells to respond to antigens and mitogens is reduced. The effect on macrophages is particularly marked and limits their ability to phagocytose and kill microorganisms and to produce tumor necrosis factor-, interleukin-1, metalloproteinases, and plasminogen activator. Both macrophages and lymphocytes produce less interleukin-12 and interferon-, important inducers of TH1 cell activity, and cellular immunity.
In addition to their effects on leukocyte function, glucocorticoids influence the inflammatory response by reducing the prostaglandin, leukotriene, and platelet-activating factor synthesis that results from activation of phospholipase A2. Finally, glucocorticoids reduce expression of cyclooxygenase-2, the inducible form of this enzyme, in inflammatory cells, thus reducing the amount of enzyme available to produce prostaglandins.
Glucocorticoids cause vasoconstriction when applied directly to the skin, possibly by suppressing mast cell degranulation. They also decrease capillary permeability by reducing the amount of histamine released by basophils and mast cells.
The anti-inflammatory and immunosuppressive effects of glucocorticoids are largely due to the actions described above. In humans, complement activation is unaltered, but its effects are inhibited. Antibody production can be reduced by large doses of steroids, though it is unaffected by moderate dosages (eg, 20 mg/d of prednisone).
The anti-inflammatory and immunosuppressive effects of these agents are widely useful therapeutically but are also responsible for some of their most serious adverse effects.
F. OTHER EFFECTS
Glucocorticoids have important effects on the nervous system. Adrenal insufficiency causes marked slowing of the alpha rhythm of the electroencephalogram and is associated with depression. Increased amounts of glucocorticoids often produce behavioral disturbances in humans: initially insomnia and euphoria and subsequently depression. Large doses of glucocorticoids may increase intracranial pressure (pseudotumor cerebri).
Glucocorticoids given chronically suppress the pituitary release of ACTH, growth hormone, thyroid-stimulating hormone, and luteinizing hormone.
Large doses of glucocorticoids have been associated with the development of peptic ulcer, possibly by suppressing the local immune response against Helicobacter pylori. They also promote fat redistribution in the body, with increase of visceral, facial, nuchal, and supraclavicular fat, and they appear to antagonize the effect of vitamin D on calcium absorption. The glucocorticoids also have important effects on the hematopoietic system. In addition to their effects on leukocytes described above, they increase the number of platelets and red blood cells.
In the absence of physiologic amounts of cortisol, renal function (particularly glomerular filtration) is impaired, vasopressin secretion is augmented, and there is an inability to excrete a water load normally.
Glucocorticoids have important effects on the development of the fetal lungs. Indeed, the structural and functional changes in the lungs near term, including the production of pulmonary surface-active material required for air breathing (surfactant), are stimulated by glucocorticoids.
|
|
Figure 2-6. Mechanism of glucocorticoid action. The glucocorticoid receptor polypeptide is schematically depicted as a protein with three distinct domains. A heat-shock protein, hsp90, binds to the receptor in the absence of hormone and prevents folding into the active conformation of the receptor. Binding of a hormone ligand (steroid) causes dissociation of the hsp90 stabilizer and permits conversion to the active configuration. |
|
|
|
Delivery of Glucocorticoids
Systemic glucocorticoids, although not routinely used for asthma treatment given the potential for side effects, are nonetheless still used for acute asthma exacerbations, and chronic, severe asthma. However, the development of aerosol delivery systems for glucocorticoids has led to dramatic increases in the therapeutic index of glucocorticoid treatment for less severe, chronic asthma. Thus, this allows for the generalized anti-inflammatory actions of this hormone to be exploited. Various glucocorticoid formulations are available for aerosol delivery that differ in their affinity for the glucocorticoid receptor. Various factors influence the choice and dose of the drug used including the severity of the disease and the devise used for drug delivery. However, maximal improvement of lung function may not occur until several weeks after treatment.
Several systemic effects of inhaled steroids have been described and include dermal thinning and skin capillary fragility. Inhaled steroids may have local side effects due to the deposition of inhaled steroid in the oropharynx.
The most common problems are hoarseness and dysphonia. Oropharyngeal candidiasis occurs in 5% of patients.
Even with proper use of aerosol devices, typically 10% of inhaled glucocorticoids are deposited in lung with the remainder swallowed and absorbed in the gut. Thus, even inhaled glucocorticoids have the potential to exert systemic effects.
The aerosol delivery of glucocorticoids to the lungs limits systemic exposure to the hormone and greatly reduces side effects.
Some new analogs of potent glucocorticoids are being used (i.e. fluticasone propionate, the active component of FLONASE) that are subjected to rapid inactivation in liver. The only circulating metabolite of fluticasone propionate detected in man is its 17ß-carboxylic acid derivative, which is formed through the cytochrome P450 3A4 pathway. This inactive metabolite had less affinity (approximately 1/2,000) than the parent drug for the glucocorticoid receptor of human lung cytosol in vitro and negligible pharmacological activity in animal studies. Thus, the bioavailability of these drugs is negligible outside of the airways. The risk of systemic effects due to improper inhalation and swallowing of the drug is dramatically reduced.
Glucocorticoids can also be combined with long-acting ß-adrenergic receptor agonists in a single inhaler devise. This can lead to an enhancement of the anti-inflammatory action of glucocorticoids at lower doses.
New generation synthetic glucocorticoids with more rapid metabolism in the liver overcome potential side effects due to ingested hormone upon aerosol delivery.
Another strategy currently being evaluated by many researchers is the development of “dissociated” glucocorticoids. These compounds are expected to unleash the gene repression activity of the glucocorticoid receptor while having little or lessened impact on the gene activation activity of the receptor. Since the anti-inflammatory actions of glucocorticoids are mainly but not exclusively due to gene repression, these compounds should still have anti-inflammatory activity but reduced side effects. The detrimental side effects of glucocorticoids are thought to be due principally to gene activation by the glucocorticoid receptor.
New generation synthetic glucocorticoids that maintain gene repression but limit gene activation by the glucocorticoid receptor (i.e. dissociated glucocorticoids) may hold promise as anti-inflammatory drugs with reduced side effects.
Glucocorticoid Resistance
Corticosteroid-dependent (CD) asthma, a situation of reduced responsiveness to glucocorticoids, is common and requires high inhaled or oral doses for disease control. In CD patients, asthma conditions worsen if glucocorticoid doses are reduced. A rare form of CD asthma is found in 1/1,000 asthma patients where complete corticosteroid resistance is observed. This rare corticosteroid-resistant (CR) asthma is defined as a failure to improve lung function by more than 15% after treatment with high doses of prednisolone (30-40 mg daily) for 2 weeks. The mechanisms responsible for CR or CD may involve disruptions in glucocorticoid receptor function. These include reduced nuclear translocation of the glucocorticoid receptor or disruptions in histone modifications in chromatin of glucocorticoid receptor regulated genes. Patients with CR asthma can be treated with long-acting inhaled ß2-agonists, as they often have a good bronchodilator response to these agents. Theophylline may also be effective for the treatment of CR asthma, but its effects are not mediated by its inhibition of phosphodiesterease. The effectiveness of theophylline in the treatment of CR asthma may be due to its action at the genome level to decrease the extent of chromatin-associated histone protein acetylation (i.e. through the increased activity of histone deacetylase enzymes).
When corticosteroids are administered from sources outside the body, they are given mainly for replacement or therapeutic purposes. Replacement involves small doses to correct a deficiency state and restore normal function. Therapeutic purposes involve relatively large doses to exert pharmacologic effects. Drug effects involve extension of the physiologic effects of endogenous corticosteroids and new effects that do not occur with small, physiologic doses. The most frequently desired effects are anti-inflammatory, immunosuppressive, antiallergic, and antistress. These are glucocorticoid effects. Mineralocorticoid and androgenic effects are usually considered adverse reactions. Additional characteristics of therapeutic corticosteroids include the following:
• All adrenal corticosteroids are available as drug preparations, as are many synthetic derivatives developed by altering the basic steroid molecule in efforts to increase therapeutic effects while minimizing adverse effects. These efforts have been most successful in decreasing mineralocorticoid activity.
• The drugs are palliative; they control many symptoms but do not cure underlying disease processes. In chronic disorders, they may enable a client to continue the usual activities of daily living and delay disability. However, the disease may continue to progress and long-term use of systemic corticosteroids inevitably produces serious adverse effects.
• Drug effects vary, so a specific effect may be considered therapeutic in one client but adverse in another. For example, an increased blood sugar level is therapeutic for the client with adrenocortical insufficiency or an islet cell adenoma of the pancreas, but an adverse reaction for most clients, especially for those with diabetes mellitus. In addition, some clients respond more favorably or experience adverse reactions more readily than others taking equivalent doses. This is partly caused by individual differences in the rate at which corticosteroids are metabolized.
• Administration of exogenous corticosteroids suppressesmthe HPA axis. This decreases secretion of corticotropin, which, in turn, causes atrophy of the adrenal cortex and decreased production of endogenous adrenal corticosteroids. Daily administration of physiologic doses (15 to 20 mg of hydrocortisone or its equivalent) or administration of pharmacologic doses (more than 15 to 20 mg of hydrocortisone or its equivalent) for approximately 2 weeks suppresses the HPA axis. HPA recovery usually occurs within a few weeks or months after corticosteroids are discontinued, but may take 9 to 12 months. During that time, supplemental corticosteroids are usually needed during stressful situations (eg, fever, illness, surgical procedures) to improve the client’s ability to respond to stress and prevent acute adrenocortical insufficiency.
• Hydrocortisone, the exogenous equivalent of endogenous cortisol, is the prototype of corticosteroid drugs. When a new corticosteroid is developed, it is compared with hydrocortisone to determine its potency in producing anti-inflammatory and antiallergic responses, increasing deposition of liver glycogen, and suppressing secretion of corticotropin.
• Anti-inflammatory activity of glucocorticoids is approximately equal when the drugs are given in equivalent doses (hydrocortisone 20 mg; prednisone and prednisolone 5 mg; methylprednisolone and triamcinolone 4 mg; dexamethasone 0.75 mg; and betamethasone
0.6 mg). Mineralocorticoid activity is high in cortisone (which is rarely used), intermediate in hydrocortisone, prednisolone, and prednisone, and low in newer agents.
• Many glucocorticoids are available for use in different clinical problems, and routes of administration vary. Several of these drugs can be given by more than one route; others can be given only orally or topically. For example, in recent years there have been several formulations developed for oral inhalation in the treatment of asthma and for nasal inhalation in the treatment of allergic rhinitis.
For intramuscular or intravenous injections, sodium phosphate or sodium succinate salts are used because they are most soluble in water. For intra-articular or intralesional injections, acetate salts are used because they have low solubility in water and provide prolonged local action.
• Duration of action also varies and is only known for oral drugs. Betamethasone and dexamethasone last 48 hours, methylprednisolone, prednisolone, prednisone and triamcinolone last 18 to 36 hours, and hydrocortisone lasts 18 hours.
INDICATIONS FOR USE
Corticosteroids are extensively used to treat many different disorders. Except for replacement therapy in deficiency states, the use of corticosteroids is largely empiric. Because the drugs affect virtually every aspect of inflammatory and immune responses, they are used in the treatment of a broad spectrum of diseases with an inflammatory or immunologic component.
These disorders include the following:
• Allergic or hypersensitivity disorders, such as allergic reactions to drugs, serum and blood transfusions, and dermatoses with an allergic component
• Collagen disorders, such as systemic lupus erythematosus, scleroderma, and periarteritis nodosa. Collagen is the basic structural protein of connective tissue, tendons, cartilage, and bone, and it is therefore present in almost all body tissues and organ systems. The collagen disorders are characterized by inflammation of various body tissues. Signs and symptoms depend on which body tissues or organs are affected and the severity of the inflammatory process.
• Dermatologic disorders that may be treated with systemic corticosteroids include acute contact dermatitis, erythema multiforme, herpes zoster (prophylaxis of postherpetic neuralgia), lichen planus, pemphigus, skin rashes caused by drugs, and toxic epidermal necrolysis.
• Endocrine disorders, such as adrenocortical insufficiency and congenital adrenal hyperplasia. Corticosteroids are given to replace or substitute for the natural hormones (both glucocorticoids and mineralocorticoids) in cases of insufficiency and to suppress corticotropin when excess secretion causes adrenal hyperplasia. These conditions are rare and account for a small percentage of corticosteroid usage.
• Gastrointestinal disorders, such as ulcerative colitis and regional enteritis (Crohn’s disease)
• Hematologic disorders, such as idiopathic thrombocytopenic purpura or acquired hemolytic anemia
• Hepatic disorders characterized by edema, such as cirrhosis and ascites
• Neoplastic disease, such as acute and chronic leukemias, Hodgkin’s disease, other lymphomas, and multiple myeloma. The effectiveness of corticosteroids in these conditions probably stems from their ability to suppress lymphocytes and other lymphoid tissue.
• Neurologic conditions, such as cerebral edema, brain tumor, and myasthenia gravis
• Ophthalmic disorders, such as optic neuritis, sympathetic ophthalmia, and chorioretinitis
• Organ or tissue transplants and grafts (eg, kidney, heart, bone marrow). Corticosteroids suppress cellular and humoral immune responses and help prevent rejection of transplanted tissue. Drug therapy is usually continued as long as the transplanted tissue is in place.
• Renal disorders characterized by edema, such as the nephrotic syndrome
• Respiratory disorders, such as asthma, status asthmaticus, chronic obstructive pulmonary disease (COPD), and inflammatory disorders of nasal mucosa (rhinitis). In asthma, corticosteroids increase the number of betaadrenergic receptors and increase or restore responsiveness of beta receptors to beta-adrenergic bronchodilating drugs. In asthma, COPD, and rhinitis, the drugs decrease mucus secretion and inflammation.
• Rheumatic disorders, such as ankylosing spondylitis, acute and chronic bursitis, acute gouty arthritis, rheumatoid arthritis, and osteoarthritis
• Shock. Corticosteroids are clearly indicated only for shock resulting from adrenocortical insufficiency (addisonian or adrenal crisis), which may mimic hypovolemic or septic shock. Studies indicate that the drugs are not beneficial in treating septic shock. In anaphylactic shock resulting from an allergic reaction, corticosteroids may increase or restore cardiovascular responsiveness to adrenergic drugs.
Contraindications to Use
Corticosteroids are contraindicated in systemic fungal infections and in people who are hypersensitive to drug formulations. They should be used with caution in clients at risk for infections (they may decrease resistance), clients with infections (they may mask signs and symptoms so that infections become more severe before they are recognized and treated), diabetes mellitus (they cause or increase hyperglycemia), peptic ulcer disease, inflammatory bowel disorders, hypertension, congestive heart failure, and renal insufficiency.
SELECTION OF DRUG DOSAGE SCHEDULE
Since these preparations differ with respect to relative anti-inflammatory and mineralocorticoid effect, duration of action, cost, and dosage forms available, these factors should be taken into account in selecting the drug to be used.
|
||||||||||||||||||||||||||||||||
|
A. ACTH VERSUS ADRENOCORTICAL STEROIDS
In patients with normal adrenals, ACTH was used to induce the endogenous production of cortisol to obtain similar effects. However, except when the increase in androgens is desirable, the use of ACTH as a therapeutic agent has been abandoned. Instances in which ACTH was claimed to be more effective than glucocorticoids were probably due to the administration of smaller amounts of corticosteroids than were produced by the dosage of ACTH.
B. DOSAGE
In determining the dosage regimen to be used, the physician must consider the seriousness of the disease, the amount of drug likely to be required to obtain the desired effect, and the duration of therapy. In some diseases, the amount required for maintenance of the desired therapeutic effect is less than the dose needed to obtain the initial effect, and the lowest possible dosage for the needed effect should be determined by gradually lowering the dose until a small increase in signs or symptoms is noted.
When it is necessary to maintain continuously elevated plasma corticosteroid levels in order to suppress ACTH, a slowly absorbed parenteral preparation or small oral doses at frequent intervals are required. The opposite situation exists with respect to the use of corticosteroids in the treatment of inflammatory and allergic disorders. The same total quantity given in a few doses may be more effective than when given in many smaller doses or in a slowly absorbed parenteral form.
Severe autoimmune conditions involving vital organs must be treated aggressively, and undertreatment is as dangerous as overtreatment. In order to minimize the deposition of immune complexes and the influx of leukocytes and macrophages, 1 mg/kg/d of prednisone in divided doses is required initially. This dose is maintained until the serious manifestations respond. The dose can then be gradually reduced.
When large doses are required for prolonged periods of time, alternate-day administration of the compound may be tried after control is achieved. When used in this manner, very large amounts (eg, 100 mg of prednisone) can sometimes be administered with less marked adverse effects because there is a recovery period between each dose. The transition to an alternate-day schedule can be made after the disease process is under control. It should be done gradually and with additional supportive measures between doses.
When selecting a drug for use in large doses, a medium- or intermediate-acting synthetic steroid with little mineralocorticoid effect is advisable. If possible, it should be given as a single morning dose.
C. SPECIAL DOSAGE FORMS
The use of local therapy, such as topical preparations for skin disease, ophthalmic forms for eye disease, intra-articular injections for joint disease, inhaled steroids for asthma, and hydrocortisone enemas for ulcerative colitis, provides a means of delivering large amounts of steroid to the diseased tissue with reduced systemic effects.
Beclomethasone dipropionate and several other glucocorticoidsprimarily budesonide and flunisolide and mometasone furoate, administered as aerosolshave been found to be effective in the treatment of asthma.
Beclomethasone dipropionate, triamcinolone acetonide, budesonide, flunisolide, and mometasone furoate are available as nasal sprays for the topical treatment of allergic rhinitis. They are effective at doses (one or two sprays one, two, or three times daily) that in most patients result in plasma levels too low to influence adrenal function or have any other systemic effects.
Corticosteroids incorporated in ointments, creams, lotions, and sprays are used extensively in dermatology.
References:
1.
2. Ray WA et al: COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet 2002;360:1071. [PMID: 12383990]
3. http://www.rheumatology.org/publications/hotline/0305NSAIDs.asp

