METHODS OF CLINICAL EXAMINATION OF ENDOCRINE GLANDS. SEMIOTICS OF HYPO- AND HYPERFUNCTION OF SOME ENDOCRINE GLANDS AND DISEASES OF THE ENDOCRINE SYSTEM. CARE OF A CHILD
The major chemical regulators of the body are the internal secretions and their secreting cells, which are collectively known as the endocrine system.
Ordinarily the endocrine system of the newbom is adequately developed, but its functions are immature. For example, the posterior lobe of the pituitary gland produces limited quantities of antidiuretic hormone (ADH), or vasopressin, which inhibits diuresis. This renders the neonate highly susceptible to dehydration.
The effect of maternal sex hormones is particularly evident in the newbom because it causes a miniature puberty. The labia are hypertrophied, and the breasts may be engorged and secrete milk during the first few days of life. Female newborns sometimes have pseudomenstruation from the sudden drop in the levels of progesterone and estrogen.
The endocrine system is adequately developed at birth, but its functions are immature. The interrelatedness of all the endocrine organs has a major effect on the function of any one gland. The lack of homeostatic control because of various functional deficiencies renders the infant especially vulnerable to imbalances in fluid and electrolytes, glucose concentration, and aminoacid metabolism.
For example, corticotropin (ACTH) is produced in limited quantities during infancy. ACTH acts on the adrenal cortices to produce their hormones, particularly the glucocorticoids and aldosterone. Because the feedback mechanism between ACTH and the adrenal cortex is immature during infancy, there is much less tolerance for stressful conditions, which affect fluid and electrolytes and the metabolism of fats, proteins, and carbohydrates. In addition, although the islets of Langerhans produce insulin and glucagon during fetal life and early infancy, blood sugar levels tend to remain labile, particularly under conditions of stress.
The function of the endocrine system is:
– to secrete intracellularly synthesized hormones into the circulation ,
– to serve as pacemaker substances for metabolic processes,
– together with the closely related but more rapidly reacting nervous system,
– to serve to integrate the various physiologic functions of the organism in adjusting to external and internal environmental demands.
Endocrine substances even in extremely small concentrations are effective in modifying metablism, behavior, and development.
The endocrine system consists of three components:
· the cell, which sends a chemical message by means of a hormone;
· the target cells, or end organs, which receive the chemical message;
· the environment through which the chemical is transported (blood, lymph, extracellular fluids) from the site of synthesis to the sites of cellular action.
Some hormones, such as acetylcholine, have specific local effects; others are secreted by specific endocrine glands and then transported by the fluids to create their effects on target tissues at locations distant from the secreting glands. Some of the general hormones, such as thyroid hormone and growth hormone, affect most cells of the body, whereas the effect of others, such as the tropic hormones, is chiefly restricted to some specific tissues.

Neuroendocrine interrelationships
Homeostasis is maintained by two regulatory systems: the endocrine and the autonomic nervous systems (also called collectively the neuroendocrine system). The endocrine system traditionally consists of seven glands located throughout the body. Three additional structures are also considered endocrine glands, although for the following reasons they are not usually included. The functions of the pineal body (epiphysis cerebri), which is located in the cranial cavity behind the midbrain and third ventricle, are largely speculative. The thymus, located behind the sternum and below the thyroid gland, plays an important role in immunity, but only during fetal life and early childhood. The placenta, which secretes ovarian hormones and chorionic gonadotropin, is only a temporary endocrine gland. The endocrine glands secrete chemicals known as hormones directly into the bloodstream. Because the glands have no ducts, they are sometimes called ductless glands, in contrast to exocrine, or duct, glands.
The autonomic nervous system consists of the sympathetic and parasympathetic systems. It controls nonvoluntary functions, specifically of smooth muscle, myocardium, and glands. The parasympathetic system is primarily involved in regulating digestive processes, whereas the sympathetic system functions to maintain homeostasis during stress. The higher autonomic centers, located in the hypothalamus and limbic system, help control both sympathetic and parasympathetic functioning. The autonomic chemical transmitters are acetylcholine, released by cholinergic fibers, and nor-epinephrine, released by adrenergic fibers. Neural release of norpinephrine into the plasma produces the same effects as secretion of this substance by the adrenal medulla. This similarity in chemical activity demonstrates the interrelatedness between the two systems.
The neuroendocrine system acts by synthesizing and releasing various chemical substances that regulate body functions. Information is carried by means of neural impulses in the autonomic system and by the blood in the endocrine system. In general, neural responses are more rapid and localized; endocrine responses are more lasting and widespread. The two systems function synergistically because neural impulses transmitted to the central nervous system stimulate the hypothalamus to manufacture and release several releasing or inhibiting factors. These substances are transferred to the anterior pituitary gland, where they lead to the release of certain tropic hormones.
Control of the endocrine system
The endocrine system controls or regulates metabolic processes governing energy production, growth, fluid and electrolyte balance, response to stress, and sexual reproduction. Hormones (chemical transmitters) are released by the endocrine gland into the bloodstream, in which they are carried to tissues that are responsive to them (target cells). The target may be another endocrine gland or an organ or tissue. Regulation of hormonal control is based on a feedback system. Usually the feedback control is one of negative funcdon, which means that an increase in one hormone results in a decrease in another substance.
The main endocrine gland controlling the release of other hormones is the pituitary gland (hypophysis). For this reason it is often called the “master gland.” The anterior lobe of the pituitary secretes tropic (which literally means “turning”) hormones that regulate the secretion of hormones from various target organs. Decreased levels of target cell hormones result in increased secretion of tropic hormones. As blood concentrations of the target hormones reach normal levels, a negative message is sent to the anterior pituitary to inhibit its production of the tropic hormone. For example, thyroid-stimulating hormone (TSH) responds to low levels of circulating thyroid hormone (TH). As blood levels of thyroid hormone reach normal concentrations, a negative feedback message is sent to the anterior pituitary, resulting in a diminished release of thyroid-stimulating hormone.
The pituitary gland is under the influence of the hypothalamus. Especially in times of stress, the hypothalamus receives messages from the central nervous system that result in the synthesis and secretion of certain hypothalamic chemicals called neurosecretions or releasing factors. These chemicals are transported by way of the pituitary portal system to the anterior pituitary, where they stimulate the secretion of tropic hormones. An example of this is the secretion of corticotropin-releasing factor (CRF) by the hypothalamus, which stimulates the pituitary to secrete adrenocorti-cotropic hormone (ACTH). In this instance the anterior pituitary is the target of the hypothalamus and secondarily effects a response from another target gland, the adrenals. The adrenals in turn secrete glucocorticoids, which have multiple target sites throughout the body.
Not all hormones are dependent on other hormones for their release. For example, insulin production depends on blood glucose concentrations. Other hormones not under the control of the pituitary gland are glucagon, parathyroid hormone (PTH), antidiuretic hormone (ADH), and aldosterone.
Because of the interdependent relationship of these glands, a malfunction in one gland produces effects elsewhere in the body. Endocrine dysfunction may result because of an intrinsic defect in the target gland (primary) or because of a diminished or elevated level of tropic hormones (secondary).
Endocrine problems occur from hypofunction or hyperfunction of the glands. Primary hypofunction is usually associated with a more profound deficiency of the target gland hormone because little or no hormone is secreted. In secondary dysfunction the target glands secrete some of their hormones but in smaller amounts and less rapidly. Hyperfunction may be the result of an increase in the tropic hormones (primary) with a consequent increase in the target gland hormones (secondary) or a hypersecretion of the target glands.
The major hormones that promote physical growth are thyroid hormone, growth hormone, and sex hormones. Insulin can be said to promote growth by its effect on carbohydrate metabolism, whereas cortisol inhibits growth. Therefore, deficiencies of growth-promoting hormones or an excess of cortisol can cause growth retardation in children. Endocrine deficiencies can be the result of abnormal secretory function in the glands responsible for their production, the pituitary hormones that stimulate their secretion; or the releasing factors from the hypothalamus. In some instances growth retardation may be the result of increased production of factors that inhibit hormone secretion.
Fig. Anterior pituitary hormones and their organs. Tropic hormones: ACTH (adrenocortico-tropic hormone); TSH (thyroid-stimulating hormone); FSH (follicle-stimulating hormone); LH (luteinizing hormone); ICSH (male analogue of LH); MSH (melanocyte-stimulating hormone); GH (STH) (growth hormone).
The thyroid gland.
The thyroid gland is an unpaired gland, locaed in the anterior part of the neck (to the front and lateral to trachea). It consists of two lobes (right and left) differing in size which more often is connected among themselves by an unpaired isthmus. The fetal thyroid bilobed shape is recognized by 7 wk of gestation, and characteristic thyroid follicle cell and colloid formation is seen by 10 wk. Thyroglobulin synthesis occurs from 4 wk, iodine trapping occurs by 8-10 wk, and thyroxine (T4) and, to a lesser extent, triiodothyronine (T3) synthesis and secretion occur from 12 wk of gestation. Hypothalamic neurons synthesize thyrotropin-releasing hormone (TRH) by 6-8 wk, the pituitary portal vessel system begins development by 8-10 wk, and thyroid-stimulating hormone (TSH) secretion is evident by 12 wk of gestation. Maturation of the hypothalamic-pituitary-thyroid axis occurs over the 2nd half of gestation, but normal feedback relationships are not mature until approximately 3 mo of postnatal life.
Thyroid and parathyroid glands.
The main function of the thyroid gland is to synthesize T4 and T3. Thyroid hormones increase oxygen consumption, stimulate protein synthesis, influence growth and differentiation, and affect carbohydrate, lipid, and vitamin metabolism. The free hormones enter cells, where T4 may be converted to T3 by deiodination. Function of thyroid hormons (video).
The thyroid is regulated by TSH, a glycoprotein produced and secreted by the anterior pituitary. This hormone activates adenylate cyclase in the thyroid gland and is important in all steps of thyroid hormone biosynthesis, from trapping of iodine to release of thyroid hormones. TSH synthesis and release are stimulated by TSH-releasing hormone (TRH), which is synthesized in the hypothalamus and secreted into the pituitary. TRH is found in other parts of the brain besides the hypothalamus and in many other organs; aside from its endocrine function, it may be a neurotransmitter. In states of decreased production of thyroid hormone, TSH and TRH are increased. Exogenous thyroid hormone or increased thyroid hormone synthesis inhibits TSH and TRH production. Except in the neonate, levels of TRH in serum are very low. Thyroid hormone physiology (video).
Further control of the level of circulating thyroid hormones occurs in the periphery. In many nonthyroidal illnesses, extrathyroidal production of T3 decreases; factors that inhibit thyroxine-type I 5′-deiodinase include fasting, chronic malnutrition, acute illness, and certain drugs. Levels of T3 may be significantly decreased, whereas levels of free T4 and TSH remain normal. Presumably, the decreased levels of T3 result in decreased rates of oxygen production, of substrate use, and of other catabolic processes. Thyroid physiology and regulation (video).
Examination of thyroid gland.
Examination of the thyroid gland (vIdeo). The thyroid is one of the endocrinal glands which is easily palpable.
Palpation of thyroid gland (video).
With the patient sitting, the thyroid gland should be carefully inspected, auscultated, and palpated during the examination of the anterior aspect of the neck. Is helpful to have a glass of water available to test swallowing several times. Keep in mind that the thyroid may be congenitally located as high as in the sublingual area or as low as in a substernal position. If the latter is the case, percussion over the upper sternum will reveal a flat tone rather than the normal resonant sound.
The thyroid may be palpated from either the back of the patient with bimanual palpation or at each side of the patient using one active hand and the other offering counterpressure. When the examiner is behind the patient, the examiner’s fingertips are placed bilaterally on each side of the Adam’s apple as an initial landmark. Drop the fingers slightly below the cricoid cartilage where the thyroid isthmus crosses the midline. Have the patient swallow and check the normal upward movement. With the patient taking small slips of water, search laterally and cephalad to determine if the upper lobes can be delineated and if their consistency, size, and contour can be appreciated. A normal thyroid feels smooth and “fleshy” to the touch as would a normal muscle. Increased firmness will be noted upon inflammation or an early neoplastic process. A bruit may be heard over the gland in hyperthyroid states. The gland is usually not palpable in hypothyroidism; in thyroiditis, the pyramidal lobe on the isthmus is usually prominent and painful when palpated.
Criteria of normal thyroid glands at palpation are:
· Painfulness – is absent.
· Surface – is smooth.
· Consistency – is soft.
· Mobility – is mobile (during swallowing movements).
· Pulsation – is absent.
Thyroid hormone studies.
Serum Thyroid Hormones.
Methods are available to measure all the thyroid hormones in serum: T4, free T4, T3, and free T3. A metabolically inert T3 (3,5′,3′-triiodothyronine), called reverse T3, is also present in serum. Age must be considered in interpreting results, particularly in the neonate.
Thyroglobulin is a glycoprotein dimer that is secreted through the apical surface of the thyrocyte into the colloid. Small amounts escape into the circulation and are measurable in serum. Levels increase with TSH (also called thyrotropin) stimulation and decrease with TSH suppression. Thyroglobulin levels are increased in the neonate, in patients with
TSH levels in serum are an extremely sensitive indicator of primary hypothyroidism. A 3rd generation of assays (chemiluminescent assays) that can measure complete suppression of TSH below the normal range is standard. These sensitive TSH assays obviate the need for TRH stimulation in the diagnosis of most patients with thyroid disorders.
In Vivo Radionuclide Studies.
Markedly improved direct tests of thyroid function have made radioiodine uptake studies less necessary. The iodine trapping or concentrating mechanism of the thyroid can be evaluated by measuring the uptake of radioactive isotope 123I (half-life, 13 hr). The technology allows doses of radioiodine (0.1-0.5 mCi) that are only a fraction of those formerly used with 131I. Technetium (99mTc) is a particularly useful radioisotope for children because in contrast to iodine, it is trapped but not organified by the thyroid and has a half-life of only 6 hr.
Thyroid ultrasonographic studies.
Thyroid ultrasound examinations can determine the location, size, and shape of the thyroid gland, and they can assess the solid or cystic nature of nodules. Ultrasound is not as reliable as radionuclide studies in evaluating infants with suspected thyroid dysgenesis, particularly ectopic glands. Ultrasound examinations are useful in identifying normal thyroid gland position in children with suspected thyroglossal duct cysts. In children with autoimmune thyroiditis, ultrasound reveals scattered hypoechogenicity. Ultrasound examinations are more accurate than physical examination in estimating goiter size and assessing thyroid nodules.
Diseases and conditions of the thyroid gland.
The basic diseases and conditions of the thyroid gland are:
1. Endemic goiter (goiter is an increase in size of thyroid gland of miscellaneous genesis) – most commonly seen in areas with insufficient supply of iodine which in tern gives rise to iodine insufficiency in the body of an organism. Eternally characterized by increase in size of the gland, more often without disorders of its function.
2. Grave’s disease (hyperthyroidism, exophtalmic/toxic goiter, Basedow’s disease, Parry’s disease) is an increase of thyroid gland with its hyperfunction. In this pathological condition there will be disorders in metabolism and functions of different organs and systems.
3. Hypothyroidism – is the pathological condition with decrease in function of the thyroid gland.
4. Thyrotoxicosis – toxic condition due to hyperfunction of the thyroid gland.
5. Euthyroid – a condition with normal thyroid gland function.
Thyroid hormone deficiency. Thyroid hormone deficiency is always associated with poor growth and delayed bone maturation. Hypothyroidism that is present from birth causes severe stunting of linear growth, which is evident early in life. When the deficiency begins before the skeletal age of 9 or 10 years, the child maintains infantile proportions with short legs compared to the length of the spine; he tends to be pale, sluggish, inactive, and obese; and intellectual achievement at school deteriorates. Acquired hypothyroidism varies with the degree and duration of the deficiency, but skeletal age is delayed if the condition has been present more than 12 months.
Congenital hypothyroidism (cretinism)
Cretinism is usually caused by failure of embryonic development of the thyroid gland, but it may also be a result of inborn enzymatic defects in the synthesis of thyroxine. The severity of the disorder depends on the amount of thyroid tissue present. Usually the neonate does not exhibit obvious signs of hypothyroidism, probably because of the exogenous source of thyroid hormone supplied by means of the maternal circulation. Manifestations are delayed in breast-fed infants. In another type of cretinism, transfer of goitrogens (substances that can induce a goiter), such as the antithyroid drugs phenylbutazone, paraaminosalicylic acid, and cobalt, may inhibit thyroid secretion, thereby resulting in congenital cretinism. Although the latter is self-limiting, it is a potentially fatal condition because once the maternal supply is terminated the infant’s thyroid is unable to produce its own hormones. In addition, a large goiter in a neonate may cause total obstruction of the airway.
Clinical manifestations. The symptoms of cretinism usually become apparent by 3 to 6 months of age in bottle-fed infants. However, before this time the earliest symptoms indicating hypothyroidism include prolonged physiologic jaundice, feeding difficulties, inactivity (excessive sleeping and minimal crying), anemia, and problems resulting from hypotonic abdominal musculature, such as constipation, diastasis recti, protruding abdomen, and umbilical hernia. The behavioral characteristics often lead parents to describe the infant as exceptionally “quiet and good.”
Impaired development of the nervous system leads to mental retardation. The severity of the intellectual deficit is related to the degree of hypothyroidism and the duration of the condition before treatment. Other nervous system manifestations include slow, awkward movements, somnolence, lethargy, and abnormal deep tendon reflexes (often referred to as “hung-up” because the relaxation phase after the contraction is slow).
Because skeletal growth is severely stunted, the child is short. Unlike pituitary dwarfism, infantile proportions persist in that the length of the trunk remains long in relation to the legs. The decreased metabolic rate results in weight gain and often leads to obesity. Characteristic infantile facial features from myxedema include a short forehead, wide, puffy eyes, wrinkled eyelids, broad, short upturned nose, and a large protruding tongue. The hair is often dry, brittle, or lusterless and follows a low hairline. Dentition is delayed and usually defective. Such facial features give the child a characteristic dull exnression- The skin is yellowish from carotenemia as a result of the depressed hepatic conversion of carotene to vitamin A. Loss of heat from reduced metabolism is reflected in a cool skin. Cold intolerance is another common consequence. Anemia results in pallor, fatigue, and lethargy, and vitamin A deficiency causes thickened, coarse, dry, scaly skin.
The cardiovascular changes are slow pulse, decreased circulation, mottling, and decreased pulse pressure. The decreased cardiac rate and output are directly related to the decreased oxygen requirements from a low metabolic rate. Respiratory changes include exertional dyspnea and decreased respiratory effort.
In breast-fed infants the clinical manifestations may be delayed until the child is weaned, at which time the facial features, skin and hair changes, growth retardation, muscular hypotonia, and cardiovascular alterations become evident. Because breast milk contains suboptimum amounts of thyroid hormone, bone age is greatly retarded, usually comparable to that of a newborn. Significantly, however, intellectual functioning remains near normal.
Figure 1. 6 week old female with symptoms of jaundice due to hypothyrodism. This patient was treated with supplemental thyroid hormonal therapy, and appeared to be a normal healthy child at 1 year of age.
Figure 2. Infant with congenital hypothyroidism. A – 3 month old infant with untreated CH; picture demonstrates hypotonic posture, myxedematous facies, macroglossia, and umbilical hernia. B – Same infant, close up of face, showing myxedematous facies, macroglossia, and skin mottling. C – Same infant, close up showing abdominal distension and umbilical hernia.
Diagnostic evaluation. Several tests are available to assess thyroid activity.
· measurement of protein-bound iodine (PBI),
· measurement of free thyroxine,
· measurement of thyroid-stimulating hormone,
· measurement of thyrotropin-relefsing factor, radioimmunoassay of thyroxine and triiodothyronine.
These tests measure the amount of thyroid hormone secreted and the intactness of the homeostatic mechanisms.
Therapeutic management. Treatment involves indefinite replacement therapy with desiccated thyroid to abolish all signs of hypothyroidism and reestablish normal physical and mental development. If adequate thyroid hormone replacement is begun before 3 months of age, the chance for completely normal growth is possible and the chance for a normal intelligence quotient is increased. To avoid the risk of overdosage of thyroid hormones, regular evaluations of thyroxine and triiodothyronine levels should be assessed. Bone age surveys are also done to ensure optimum growth.
Parathyroid glands.
There are two pairs of parathyroid glands. They are adjoined to the back surface of thyroid gland. Upper pair is located between top third of thyroid gland and lower pair is located at the lower third of thyroid gland.
The basic function of parathyroid glands is the secretion of parathyroid hormone. Actions of parathyroid hormone and calcitonin are antagonistic and inter related. If calcitonin reduces the amount of calcium in blood, parathyroid hormone on the contrary, increases it. Calcitonin and parathyroid hormone simultaneously with vitamin D maintain the optimum amount of calcium and phosphorus in the organism (in blood, extracellular fluid and cell).
Figure 3. Calcium regulation in the human body. The role of parathyroid hormone is shown in blue.
Functions of parathyroid hormone are:
· Stimulates transformation of inactive vitamin D to the active form, which rases reabsorbtion of calcium in intestine.
· In bones activates process of reabsorbtion of calcium.
· In kidneys raises the reabsorbtion of calcium.
All the three specified functions raise the amount of calcium inblood.
· In kidneys reduces the reabsorbtion of phosphorus in proximal and distal canals of nephron – so the amount of phosphorus in blood decreases.
Symptomatological disorders.
1. Hyperparathyroidism – is the hyperfunction of parathyroid glands, in children it is rarely seen. The disease of newborn can be of congenital or heredirary character.
2. Hypoparathyroidism – is decreased function of the parathyroid glands with underproduction of parathyroid hormone. This can lead to low levels of calcium in the blood, often causing cramping and twitching of muscles or tetany (involuntary muscle contraction), and several other symptoms. The condition can be inherited, but it is also encountered after thyroid or parathyroid gland surgery, and it can be caused by immune system-related damage as well as a number of rarer causes. The main symptoms of hypoparathyroidism are the result of the low blood calcium level, which interferes with normal muscle contraction and nerve conduction. Hypoparathyroidism – mechanism of disease (video).
Adrenal glands.
Adrenal glands consist of two different layers, which secrete different hormones.
The cortical layer secretes:
1. Corticosteroids. Secretion of corticosteroids is characterized by every day reccurence, which develops at the 15-20th day after birth – the maximum amount is released in the morning. It is taken into account by doctors while prescribing hormonal medicines to the child. Corticosteroids are divided into two groups:
· Glucocorticoids: main of which are corticosterone and cortisone.
· Mineralocorticoids, main of which is – aldosterone.
2. Male sex hormones – androgens and female sex hormones – estrogenes. Influence in thedevelopment of secondary sexual characters.
The inner medulla layer produces catecholamines, which react on stress stimulus already in early neonatal period. They are:
· Adrenaline
· Noradrenaline
· Dopamine
Function of adrenal glands (video).
Symptomatological disorders.
One of the pathological conditions in older children is Addison’s disease, in which there is bilateral defect of cortex of adrenal glands and exlusion or reduction of gland’s hormones. External appearance of disease – significant pigmentation of the skin: color golden-brown, muddy-brown, tanned color.
Sex glands (gonades).
They produce such hormones:
· Progesterone
· Estradiol
· Estrone
· Testosterone
Synthesis of sexual hormones first of all is regulated by gonadotrophic hormones of hypophysis.
Sex hormone deficiency. Sex hormone deficiency that causes delayed puberty can occur as a result either of pituitary dysfunction or of hypogonadism. A hypofunctioning pituitary gland, as briefly discussed in the preceding segment on endocrine dysfunction, can produce a deficiency in either the gonadotropic hormones, which retards maturation of the gonads, or growth hormone, which will diminish total growth during childhood. In addition, there are a large variety of disorders that cause absence or deficiency of sex hormone secretion by their effect on the gonads directly. These may be genital abnormalities that are related to defective gonadal differentiation or those that are associated with functional abnormalities of the already differentiated fetal gonad. The largest group of disorders in which deficient gonadal development is a prominent feature includes the sex chromosomal aberrations, e.g. Klinefelter’s and Turner’s syndromes.
Syndromes of primary gonadal failure. The most frequently seen disorders associated with primary gonadal failure are the sex chromosomal defects categorized collectively as gonadal dysgenesis, principally Turner’s syndrome. Chromosomal impairment of male sexual function is most commonly caused by Klinefelter’s syndrome. Derangements that become apparent at puberty are more common. Clinical presentation in the female may be masculinization, sexual infantilism or hypoplasia, primary absence of menstruation (amenorrhea), or abnormally scanty or infrequent menstruation (oligomenorrhea or hypomenorrhea).
Gonadotropic hormones.
The gonadotropic hormones follicle-stimulating hormone, luteinizing hormone, interstitial cell-stimulating hormone, and prolactin are responsible for the growth and maturation of the gonads at puberty and for the ongoing stimulation of germ cell production during adulthood. The function of these hormones is discussed in relation to puberty and will not be elaborated here.
Sexual development of girls
Development of mammary gland
|
|
Phase |
|
Mammary glands does not project over the surface thorax |
Ма 0
|
|
Glands something project,alveolla together with nipple form uniform cone |
Ма 1
|
|
Glands much project, together with nipple and alveola have cone shape |
Ма 2
|
|
Glands body adopt roundinsh form, nipples rise over the alveola |
Ма 3
|
Grows of pubic hair
|
|
Phase |
|
Absence of hair |
P 0 |
|
Sporadic hair |
P 1 |
|
The hair on the antral part of pubic are few and long |
P 2 |
|
Hair on surface of pubic is long ,curly, dense (thick) |
P 3 |
Development of hair in axillary fossa
|
|
Phase |
|
Absence of hair |
Ах 0 |
|
Sporadic hair |
Ах 1 |
|
Hair is scarce on the central part fossa |
Ах 2 |
|
Hair is dense,curly on the entire surface of fossa |
Ах 3 |
Formation of menstrual function
|
|
Phase |
|
Absense of menstrual cycle (menses) |
Ме 0 |
|
1-2 menstrual cycle before examination |
Ме 1 |
|
Irregular menstrual cycle (menses) |
Ме 2 |
|
Regular menstrual cycle (menses) |
Ме 3 |
Sexual development of boys
Development of hair in axillary fossa
|
|
Phase |
|
Absence of hair |
Ах 0 |
|
Sporadic hair |
Ах 1 |
|
Hair is scarce on the central part |
Ах 2 |
|
Dense straight hair on the entire fossa |
Ах 3 |
|
Dense curly hair on the entire fossa |
Ах 4 |
Grows of pubic hair
|
|
Phase |
|
Absence of hair |
P 0 |
|
Sporadic hair |
P 1 |
|
Sparse growth of long, straight, downy and slightly pigmented hair at base of penis |
P 2 |
|
Hair darker, coarser and curly and spread sparsely over entire pubis |
P 3 |
|
Pubic hair more abundant with curling but restricted to pubic area |
P 4 |
|
Hair adult in quantity and type with spread to inner surface of thighs |
P 5 |
Grows of thyroid cartilage
|
|
Phase |
|
No signs of grows |
L 0 |
|
Beginning of cartilage projection |
L 1 |
|
Distinct projection of Adam’s-apple |
L 2 |
Change of voice timbre
|
|
Phase |
|
Childish voice |
V 0 |
|
Mutation (creaking)of voice |
V 1 |
|
Male timble of voice |
V 2 |
Grows of facial hair
|
|
Phase |
|
Absence of hair |
F 0 |
|
Beginning of hair grows over the upper lip |
F 1 |
|
Harsh hair over the upper lip and appearence of hair on the chin |
F 2 |
|
Spreading of hair grows over the upper lip and chin with tendency to confluence,beginning on whiskers grows |
F 3 |
|
Confluence of hair over the upper lip and chin, pronounced of whiskers grows |
F 4 |
|
Confluence of all zones of hair grows |
F 5 |
Term of sexual development
|
Age, yr |
boys |
girls |
||
|
sexual formulas |
sexual formulas |
|||
|
Since |
Up to |
Since |
Up to |
|
|
10 |
|
|
Ma0P0Ax0Me0 |
Ma2P1Ax0Me0 |
|
11 |
|
|
Ma1P1Ax0Me0 |
Ma2P1Ax0Me0 |
|
12 |
V0P0L0Ax0F0 |
V1P1L0Ax0F0 |
Ma1P0Ax0Me0 |
Ma3P3Ax1Me1 |
|
13 |
V1P0L0Ax0F0 |
V2P3L1Ax2F0 |
Ma2P2Ax0Me0 |
Ma3P3Ax2Me3 |
|
14 |
V1P2L0Ax0F0 |
V2P3L2Ax2F1 |
Ma3P2Ax2Me0 |
Ma3P3Ax3Me3 |
|
15 |
V1P4L1Ax0F0 |
V2P3L2Ax3F2 |
Ma3P3Ax2Me3 |
Ma3P3Ax3Me3 |
|
16 |
V2P4L1Ax2F1 |
V2P5L2Ax4F3 |
|
|
|
17 |
V2P2L2Ax2F0 |
V2P5L2Ax4F3
|
|
|
Diabetes mellitus
Etiology
Heredity is unquestioned as a prominent factor in the etiology of diabetes mellitus, although the mechanism of inheritance is unknown. Diabetes may be actually a syndrome rather than a specific disease. A variety of genetic mechanisms have been proposed, but most favor a multifactorial inheritance or a recessive gene somehow linked to the tissue-typing antigens, the human lymphocyte-A (HLA) system. However, the inheritance of noninsulin-dependent diabetes and insulin-dependent diabetes appears to be different. Nearly 100% of offspring of parents who both have noninsulin-dependent diabetes develop that type of diabetes, but only 45% to 60% of the offspring of both parents who have insulin-dependent diabetes will develop the disease. There is also an increased risk of diabetes with obesity. The incidence of the disease doubles with every 20% of excess weight, and this figure applies to the young as well as to the older diabetic person. Diabetes is now the sixth leading cause of death by disease in adults and the first leading cause of new cases of blindness between 20 and 75 years of age.Vimses have been implicated in the etiology of diabetes. The viral theory states that the (B-cells of some individuals (most specialists believe that the (3-cells are genetically susceptible because of the defects in the HLA system) are attacked by certain viruses, causing cell damage or death. The body reacts to this damaged or changed tissue in an autoim-mune phenomenon, forming antibodies that “attack” the (3-cells, resulting in cell death. When there are not enough available (3-cells to supply sufficient insulin to meet the needs of the body, insulin-dependent diabetes results. Tumors of the pancreas, pancreatitis, stress drugs such as steroids, stress diseases that involve other endocrine organs such as acromegaly, heredity, and viral diseases are now believed to play a part in causing diabetes.
Ioninsulin-dependent, or type II, diabetes disturbed carbohydrate metabolism may be a result of a sluggish or insensitive secretory response in the pancreas or a defect in body tissues that requires unusual amounts of insulin, or the insulin secreted may be rapidly destroyed, inhibited, or in-activated in affected persons. A lack of insulin because of reduction in islet cell mass or destruction of the islets is the hallmark of the person with insulin-dependent, or type I, diabetes.
Pathophysiology
Insulin is needed to support the metabolism of carbohydrates, fats, and proteins, primarily by facilitating the entry of these substances into the cell. Insulin is needed for the entry of glucose into the muscle and fat cells, for the prevention of mobilization of fats from fat cells, and for storage of glucose as glycogen in the cells of liver and muscle. Insulin is not needed for the entry of glucose into nerve cells or vascular tissue. The chemical composition and molecular structure of insulin are such that it fits into receptor sites on the cell membrane. Here it initiates a sequence of poorly defined chemical reactions that alter the cell membrane to facilitate the entry of glucose into the cell and stimulate enzymatic systems outside the cell that metabolize the glucose for energy production.
With a deficiency of insulin, glucose is unable to enter the cell and its concentration in the bloodstream increases. The increased concentration of glucose (hyperglycemia) produces an osmotic gradient that causes the movement of body fluid from the intracellular space to the extracellular tion in the glomerular filtrate exceeds the threshold (180 mg/ dl), glucose “spills” into the urine along with an osmotic diversion of water (polyuria), a cardinal sign of diabetes. The urinary fluid losses cause the excessive thirst (polydipsia) observed in diabetes. As might be expected, this water washout results in a depletion of other essential chemicals.
Ketoacidosis. When insulin is deficient, glucose is unavailable for cellular metabolism and the body chooses alternate sources of fuel, principally fat. Consequently fats break down into fatty acids and glycerol in the fat cells and in the liver and are converted to ketone bodies (3-hydroxy-butyric acid, acetoacetic acid, acetone). The ketone bodies are used as the alternative to glucose as a source of fuel but are utilized in the cells at a limited rate. Any excess is expelled in the urine (ketonuria) or the lungs (acetone breath).
Protein is also wasted during insulin deficiency. Since glucose is unable to enter the cells, protein is broken down and converted to glucose by the liver (glucogenesis), which glucose further contributes to the hyperglycemia. These mechanisms are similar to those seen in starvation when substrate (glucose) is absent. The body is actually in a state of starvation during insulin deficiency. Without the use of carbohydrates for energy, fat and protein stores are depleted as the body attempts to meet its energy needs. The hunger mechanism is triggered, but the increased food intake (poly-phagia) enhances the problem by further elevating the blood glucose .
Ketones are organic acids that readily produce excessive quantities of free hydrogen ions, causing a fall in plasma pH. Chemical buffers in the plasma, principally bicarbonate, combine with the hydrogen ions to form carbonic acid, which readily dissociates into water and carbon dioxide. The respiratory system attempts to eliminate the excess carbon dioxide by increased depth and rate—Kussmaul’s respirations, the hyperventilation characteristic of metabolic acidosis. The ketones are buffered by sodium and potassium in the plasma. The kidney attempts to compensate for the increased pH by increasing tubular secretion of hydrogen and ammonium ions in exchange for fixed base, thus depleting the base buffer concentration.
Potassium is also a problem and was once the cause of unexplained deaths shortly after insulin therapy was instituted. With cellular death, potassium is released from the cell into the bloodstream and excreted by the kidney where the loss is accelerated by the osmotic diuresis. The total body potassium is then decreased, even though the serum potassium level may be elevated as a result of the decreased fluid volume in which it circulates. Alteration in serum and tissue potassium can make cardiac arrest a potential problem.
If these conditions are not reversed by insulin therapy in combination with correction of the fluid deficiency and electrolyte imbalance, progressive deterioration occurs with dehydration, electrolyte imbalance, acidosis, coma, and death. Diabetic ketoacidosis should be diagnosed promptly in a seriously ill patient and therapy instituted.
Clinical manifestations
The symptomatology of diabetes is more readily recognizable in children than in adults, so it is surprising that the diagnosis may sometimes be missed or delayed. Diabetes is a great imitator: influenza, gastroenteritis, and appendicitis are the conditions most often diagnosed, only to find that the disease was really diabetes. Those families with a strong family history of diabetes should suspect diabetes, especially if there is one child in the family with diabetes.
The sequence of chemical events described previously results in hyperglycemia and acidosis, which in turn produce the three “polys” of diabetes—polyphagia, polydipsia, and polyuria—the cardinal symptoms of the disease. In nonin-sulin-dependent diabetes (which has also been found in older children), the insulin values are found to be elevated, 80% to 90% of this population have been found to be overweight, and there is often tiredness and frequent infections (such as monilial infections in females).
The insulin-dependent diabetic has markedly decreased insulin levels and, as diabetes becomes complete, there is no demonstrable insulin at all. The child may start wetting the bed, become grouchy and “not himself,” or act overly tired. Abdominal discomfort is common. Weight loss, though quite observable on the charts, may be a less frequent presenting complaint because of the fact that the family might not have noticed the change. Another outstanding feature of diabetes is thirst. One couple reported that their child, during a trip from
The child
· may be hyperglycemic, with elevated blood glucose levels and glucose in the urine;
· may be in diabetic ketosis, with ketones as well as glucose in the urine but not noticeably dehydrated;
· may be in diabetic ketoacidosis, with dehydration, electrolyte imbalance, and acidosis.
Diagnostic evaluation
Observation and testing are important to the diagnosis of diabetes in children.
If children demonstrate glycosuria, are overweight, or exhibit symptoms of hypoglycemia, they are candidates for glucose tolerance testing.
The urine test will show positive glucose only when the disease is actually manifest. A negative urine test does not necessarily rule out early diabetes, nor does a positive test necessarily indicate diabetes. Renal glycosuria, unrelated to diabetes, can result in glucose in the urine.
The fasting blood glucose test may miss the diagnosis of early diabetes and has been known to miss as many as 85 % of children who had an abnormal glucose tolerance test with asymptomatic disease. The 4-hour glucose tolerance test has been found to be the most useful test for the diagnosis of early diabetes, whereas the 6-hour glucose tolerance test is most helpful for the diagnosis of hypoglycemia. Based oorms established for normal, nondiabetic children of various ages, the criteron for the diagnosis of early diabetes is two or more abnormal tests with two or more values in each test that are outside the normal range. However, standardization of food intake before the test may be important and those preparing for the test should emphasize the importance of following the directions for diet supplied by the physician or laboratory. It is difficult to do glucose tolerance testing in children younger than 3 years of age, since norms for children in this age-group have not been established.
Problem of diagnosis. Signs, symptoms, and chemical tests may lead to the conclusion that the child has diabetes, when in reality another condition may be present. This is true in salicylate intoxication, which can be ruled out easily by boiling the urine. The acetone, if present, will boil out of the urine, leaving a negative Acetest if related to diabetes and a positive Acetest if related to salicylate intoxication. Temporary hyperglycemia may accompany such stressful conditions as bums, hyperalimentation, pancreatitis, and encephalitis. The glucose tests usually return to normal once the stress is reversed; however, insulin may be needed for a short period in the stress illnesses, especially when the child is undergoing hyperalimentation. Other abnormal conditions that may cause glucose to appear in the urine are certain renal diseases, some other endocrine disorders such as hypercortisolism, and lead encephalopathy.
Therapeutic management: insulin
The management program for any child with diabetes mellitus should involve flexibility and 24-hour insulin coverage and should be able to fit into the child’s life-style. The insulin treatment should be determined by the recognition that the effective duration of action of insulin in children may be somewhat different from that in adults. The effective action of insulin is described as the effect of a certain amount of insulin in lowering the blood glucose level over a period of time. Ideally the blood glucose level is maintained at less than 140 mg/dl and no lower than 60 mg/dl during the time of specific action of the insulin, based on past information regarding the duration of action of the intermediate-acting insulins. The accepted duration of action of intermediate-acting insulins is 24 hours or more, but in insulin-dependent children it does not appear to be the case. The duration of effective action for intermediate-acting insulin has been found to be 12 to 14 hours. Lente insulin is the longest acting of the intermediate-acting insulins, but even it lasts only 14 to 16 hours .
In working with these insulins, it is wise to remember that lente insulin is 30% semilente and 70% ultralente. Lente insulins that mix with no other insulins other than regular derive their action from the size and number of crystals—small and numerous crystals = semilente insulin;large and less numerous crystals = ultralente insulin. Pro-tamine zinc insulin (PZI) is seldom used today because of its very long duration of action and its very low tissue insulin levels, which may not saturate receptor sites on the cell membrane sufficiently well to effectively help the body utilize the glucose that may be present. The potential overlap of insulin action is unsuited for children, who are active one minute and very inactive the next. The balance that needs to be achieved between insulin, diet, and activity is most difficult when using this type of insulin. The intermediate-acting insulins (other than lente insulin) derive their delayed action from a protein tag. The most commonly used insulins are the intermediate-acting insulins, principally NPH, which are usually given in a single early morning dose combined with a small amount of short-acting inlin (usually regular).
Syndromes of endocrine system disorders
1. Syndrome of growth inhibition
2. Acromegalia, gigantism
3. Syndromes of metabolism disorders
4. Syndromes of hyperfunction of endocrine gland
5. Syndromes of hypofunction of endocrine gland
6. Syndromes of hyperglycemia
7. Syndromes of hypoglycemia
8. Syndrome of mental retardation
Congenital Hypothyroidism
Background
Congenital hypothyroidism is inadequate thyroid hormone production iewborn infants. This can occur because of an anatomic defect in the gland, an inborn error of thyroid metabolism, or iodine deficiency.
The term endemic cretinism is used to describe clusters of infants with goiter and hypothyroidism in a defined geographic area. Such areas were discovered to be low in iodine, and the cause of endemic cretinism was determined to be iodine deficiency. In the 1920s, adequate dietary intake of iodine was found to prevent endemic goiter and cretinism.Endemic goiter and cretinism are still observed in some areas, such as regions of
The term sporadic cretinism was initially used to describe the random occurrence of cretinism ionendemic areas. The cause of these abnormalities was identified as nonfunctioning or absent thyroid glands. This led to replacement of the descriptive term sporadic cretinism with the etiologic term congenital hypothyroidism. Treatment with thyroid replacement therapy was found to elicit some improvement in these infants, although many remained impaired.
An infant shown a few months after starting thyroid hormone replacement.
Infant a few months after starting thyroid hormone replacement.
The morbidity from congenital hypothyroidism can be reduced to a minimum by early diagnosis and treatment.Although initial preliminary studies were performed using thyroid-stimulating hormone (TSH) levels in cord blood,mass screening was made feasible by the development of radioimmunoassay for TSH and thyroxine (T4) from blood spots on filter paper, obtained for neonatal screening tests.
Pathophysiology
The thyroid gland develops from the buccopharyngeal cavity between 4 and 10 weeks’ gestation. The thyroid arises from the fourth branchial pouches and ultimately ends up as a bilobed organ in the neck. Errors in the formation or migration of thyroid tissue can result in thyroid aplasia, dysplasia, or ectopy. By 10-11 weeks’ gestation, the fetal thyroid is capable of producing thyroid hormone. By 18-20 weeks’ gestation, blood levels of T4 have reached term levels. The fetal pituitary-thyroid axis is believed to function independently of the maternal pituitary-thyroid axis.
The thyroid gland uses tyrosine and iodine to manufacture T4 and triiodothyronine (T3). Iodide is taken into the thyroid follicular cells by an active transport system and then oxidized to iodine by thyroid peroxidase. Organification occurs when iodine is attached to tyrosine molecules attached to thyroglobulin, forming monoiodotyrosine (MIT) and diiodotyrosine (DIT). The coupling of 2 molecules of DIT forms tetraiodothyronine (ie, T4). The coupling of one molecule of MIT and one molecule of DIT forms T3. Thyroglobulin, with T4 and T3 attached, is stored in the follicular lumen. TSH activates the enzymes needed to cleave T4 and T3 from thyroglobulin. In most situations, T4 is the primary hormone produced by and released from the thyroid gland.
Inborn errors of thyroid metabolism can result in congenital hypothyroidism in children with anatomically normal thyroid glands.
T4 is the primary thyronine produced by the thyroid gland. Only 10-40% of circulating T3 is released from the thyroid gland. The remainder is produced by monodeiodination of T4 in peripheral tissues. T3 is the primary mediator of the biologic effects of thyroid hormone and does so by interacting with a specific nuclear receptor. Receptor abnormalities can result in thyroid hormone resistance.
The major carrier proteins for circulating thyroid hormones are thyroid-binding globulin (TBG), thyroid-binding prealbumin (TBPA), and albumin. Unbound, or free, T4 accounts for only about 0.03% of circulating T4 and is the portion that is metabolically active. Infants born with low levels of TBG, as in congenital TBG deficiency, have low total T4 levels but are physiologically normal. Familial congenital TBG deficiency can occur as an X-linked recessive or autosomal recessive condition.
The contributions of maternal thyroid hormone levels to the fetus are thought to be minimal, but maternal thyroid disease can have a substantial influence on fetal and neonatal thyroid function. Immunoglobulin G (IgG) autoantibodies, as observed in autoimmune thyroiditis, can cross the placenta and inhibit thyroid function. Thioamides used to treat maternal hyperthyroidism can also block fetal thyroid hormone synthesis. Most of these effects are transient. Radioactive iodine administered to a pregnant woman can ablate the fetus’s thyroid gland permanently.
The importance of thyroid hormone to brain growth and development is demonstrated by comparing treated and untreated children with congenital hypothyroidism. Thyroid hormone is necessary for normal brain growth and myelination and for normal neuronal connections. The most critical period for the effect of thyroid hormone on brain development is the first few months of life.
Epidemiology
Frequency
The incidence of congenital hypothyroidism, as detected through newborn screening, is approximately 1 per 4000 births.An increase in the diagnosis of primary congenital hypothyroidism has been reported in New York.This trend has also been observed in some other states,although not all. Possible explanations include changing demographics of the birth population, including changes in race, ethnicity, and the incidence of low birth weight.Changes in laboratory and screening methodology may also play a role in this reported rise in incidence.Some infants identified as having primary congenital hypothyroidism may have transient disease and not permanent congenital hypothyroidism.
Twins
An increased incidence of congenital hypothyroidism is observed in twins.Twin births are approximately 12 times as likely to have congenital hypothyroidism as singletons.Usually, only one twin is hypothyroid, but a common in-utero exposure can cause hypothyroidism in both.
International
In central
Data from most countries with well-established newborn screening programs indicate an incidence of congenital hypothyroidism of about 1 per 3000-4000.Some of the highest incidences (
Although percentages of specific etiologies vary from country to country, ranges are as follows:
- Ectopic thyroid – 25-50%
- Thyroid agenesis – 20-50%
- Dyshormonogenesis – 4-15%
- Hypothalamic-pituitary dysfunction – 10-15%
Mortality/Morbidity
Congenital hypothyroidism does not affect the all-cause standardized mortality ratio in treated patients.
Profound mental retardation is the most serious effect of untreated congenital hypothyroidism. Severe impairment of linear growth and bone maturation also occurs. Affected infants whose treatment is delayed can have neurologic problems such as spasticity and gait abnormalities, dysarthria or mutism, and autistic behavior.
Race
Congenital hypothyroidism is observed in all populations. The prevalence at birth is increased in Hispanics, particularly in Hispanic females, who have a birth prevalence of
Sex
Most studies of congenital hypothyroidism suggest a female-to-male ratio of a 2:1. Devos et al showed that much of the discrepancy is accounted for by infants with thyroid ectopy.The sex ratio for Hispanics is more striking, with a 3:1 female-to-male ratio. The ratio is lower among Black infants.
Age
By definition, congenital hypothyroidism is present at, or before, birth. Children who develop primary hypothyroidism when aged 2 years or older have poor growth and slow mentation but generally do not exhibit the profound and incompletely reversible neurologic abnormalities observed in untreated congenital hypothyroidism.
Congenital Hyperinsulinism
Background
Persistent hyperinsulinemic hypoglycemia of infancy (PHHI) represents the most common cause of hyperinsulinism ieonates; currently, many authors prefer the term congenital hyperinsulinism (CHI). It was first identified in 1938, when Laidlaw coined the term nesidioblastosis to describe the neodifferentiation of islets of Langerhans from pancreatic ductal epithelium (a term since replaced by PHHI and CHI).
Severe recurrent hypoglycemia associated with an inappropriate elevation of serum insulin, C-peptide, and proinsulin levels defines CHI. If left untreated, CHI can lead to brain damage or death secondary to severe hypoglycemia. Although it was initially thought to affect only infants and children, numerous cases have been reported in adults of all ages but at a much lower incidence. CHI is often poorly responsive or unresponsive to medical management, necessitating 95% or near-total pancreatectomy.
Pathophysiology
In CHI, the histologic abnormalities in pancreatic structure are heterogeneous but can be grouped into the following 2 broad categories:
- Focal adenomatous hyperplasia (found in one fourth to one third of cases)
- Diffuse abnormality of the islets
In the focal form, the histologically abnormal beta cells are limited to 1 or more focal areas, whereas in the diffuse form, the beta-cell abnormality is distributed throughout the pancreas.
Investigations into the molecular basis of CHI have led to the discovery of mutations in the sulfonylurea receptor and an inwardly rectifying potassium channel. However, approximately 50% of cases do not involve any currently known mutation.
Presumed structural or functional molecular abnormalities in the insulin secretory mechanism or glucose-sensing mechanism result in a failure to reduce pancreatic insulin secretion in the presence of hypoglycemia (serum glucose level < 60 mg/dL). Inappropriately high circulating insulin levels act to promote hepatic and skeletal muscle glycogenesis, causing a decrease in the amount of free glucose available in the bloodstream and suppression of the formation of free fatty acid (FFA), an alternative energy substrate for the brain.
The net effect is hypoglycemia, which results in physiologically appropriate adrenergic and neuroglycopenic symptoms, with severe neurologic dysfunction and frank seizure activity when central nervous system (CNS) glucose levels fall below 20-30 mg/dL.
Prolonged hypoglycemia causes death. Repeated episodes of severe, prolonged, sublethal hypoglycemia can result in permanent neurologic damage, including developmental delay, mental retardation, and focal CNS deficits. Therapy should be aimed at prevention of hypoglycemia to prevent morbidity and mortality.
Etiology
CHI is a clinically, pathologically, and genetically heterogeneous disease. Most cases are sporadic. In approximately 50% of cases, no known genetic abnormality is found. Familial forms of CHI are rare but well documented. These cases involve autosomal recessive or dominant defects in the following 4 genes:
- Beta-cell high-affinity sulfonylurea receptor gene (ABCC8, also known as SUR1)
- Inwardly rectifying potassium channel gene (KCNJ11, also known as Kir6.2)
- Glucokinase gene (GCK, also called GK) – Only 5 persons have been described with this mutation
- Glutamate dehydrogenase gene (GLUD1, also called GUD1) – This gene is associated with hyperinsulinism with hyperammonemia; it is unclear whether GLUD1 mutation is a variant of CHI or a distinct clinical entity
Some data have helped elucidate the mechanism of the focal form of CHI. In the focal form, data have shown that a specific loss of maternal alleles occurs in the imprinted chromosome region 11p15 in the cells of the hyperplastic area, but no loss occurs in the normal pancreatic cells. This loss of heterozygosity results in a reduction to hemizygosity or homozygosity of the remaining paternal alleles that carry a mutation of ABCC8 (SUR1) or KCNJ11 (Kir6.2).
This abnormality occurs during embryonic development in a single pancreatic cell, resulting in a proliferative monoclonic lesion. However, other pancreatic cell lines not derived from this cell, as well as all other cells of the body, do not carry this genetic defect. The result is similar to uniparental disomy, but it occurs only in a clonal cell line and not constitutionally. This is a nonmendelian mechanism. This abnormality has not been observed in patients with the diffuse form of CHI.
High rates of consanguinity have beeoted in some series. No known genetic abnormalities have been found in approximately half (in some series, the majority) of the patients studied, suggesting the existence of other mutations that have not yet been described. More detailed treatments of the genetics of hyperinsulinism have been published by Glaser et aland Fournet et al.
Adult-onset hyperinsulinemic hypoglycemia with pancreatic beta cell hypertrophy has been reported in adults undergoing Roux-en-Y gastric bypass surgery.The relation between the operative procedure and the pancreatic disease remains poorly understood. Service et al theorize that gastric bypass may increase activity of beta-cell trophic factors.
Epidemiology
Few data are available on CHI. An estimated incidence of
Age- and sex-related demographics
Patients with CHI usually present between birth and age 18 months, with most cases diagnosed shortly after birth. Cases of adult-onset forms of CHI are rare but well documented.
The diffuse form of CHI has a male-to-female ratio of 1.2:1. Focal lesions are found in a 1.8:1 male-to-female ratio. The overall male-to-female ratio is 1.3:1.
Prognosis
Cure
If a solitary focal lesion can be identified and excised, the patient usually maintains blood glucose levels within the reference range without the need for medication or continuous feedings.
Hypoglycemia often persists even after a 95-98% pancreatectomy. Hypoglycemia may be easier to control after partial pancreatectomy and may resolve months or years later or persist throughout life.
In a study of 101 patients, 50% of patients who underwent a 95% or greater pancreatectomy were cured (ie, they did not require medical or dietary treatment to maintaiormoglycemia within the follow-up period of the study). The mean time from surgery to cure was 4.7 years.However, in some series, 40-63% of patients managed with medical therapy alone had late remission of hypoglycemia. Later onset of disease is correlated with a higher likelihood of being able to discontinue medical therapy.
Future development of diabetes mellitus
Patients who undergo partial pancreatectomy are at high risk for developing diabetes mellitus later in life. The risk of diabetes mellitus appears to increase with the extent of pancreatic resection; however, the risk is significant even with conservative surgical procedures.
In one series, 14% of children with diffuse lesions developed diabetes mellitus, regardless of the surgical procedure performed. The mean time from surgery to development of diabetes mellitus was 9.6 years.Because most series are limited by relatively short follow-up times, the lifetime incidence of diabetes mellitus is not well understood. Islet cell preservation and autotransplantation remain promising but untested therapies for patients who develop diabetes mellitus.
Diabetes mellitus is extremely rare after resection of focal lesions.
In a series of 3 patients treated without pancreatic resection, 2 developed impaired glucose tolerance, and one developed diabetes mellitus.All 3 patients had mutations of the ABCC8 (SUR1) gene. The significance of this small series is uncertain, but the results suggest that development of impaired glucose tolerance may be part of the underlying disease process and not solely due to surgical reduction in islet cell mass.
Education of the patient and family and long-term follow-up are essential to prevent delays in the diagnosis of disease recurrence, glucose intolerance, or diabetes mellitus.
Neurodevelopmental outcome
In some series, a high frequency of mental retardation, developmental delay, and nonhypoglycemic seizures has been observed. These findings are generally attributed to minimal brain damage from early hypoglycemic events, although the existence of these disorders as inherent comorbid conditions with CHI has not been fully excluded. Other series, usually in conjunction with medication studies, have showormal developmental progress in patients with PHHI.
Some data suggest that patients with early, severe disease treated with early, aggressive surgery have a better neurodevelopmental outcome. No comprehensive long-term studies of neurodevelopmental outcomes in patients with PHHI are available, and the heterogeneity of the disease likely confounds many neurodevelopmental studies.
Permanent neurologic dysfunction (eg, seizures, developmental delay, focal neurologic deficits) or death secondary to severe, prolonged hypoglycemia may occur if PHHI goes untreated or is inadequately treated.
Patient Education
A nutritionist should provide dietary education and meal-planning assistance. Patients (if old enough) and family members should be taught how to use a home blood glucose monitor. They should also understand the signs and symptoms of hypoglycemia and how to treat this condition with rapid-acting oral carbohydrates and subcutaneous glucagon.
Family members must understand the importance of prompt treatment of hypoglycemia to prevent severe complications or death. Family members should be instructed to call the local emergency medical service (
Patients and family members should be reminded to carry medications, a glucose meter, a rapid-acting carbohydrate source, and glucagon when traveling. Families should carry sufficient supplies for several extra days in case of unexpected travel delays.
Patients who have undergone surgery, as well as their family members, should be reminded of the risk of future development of diabetes mellitus and the importance of long-term follow-up. Failure to educate families about this potential late complication could result in a delay of diagnosis of diabetes mellitus if it occurs.
Genetic counseling with regard to risk of recurrence may be appropriate. Techniques for prenatal diagnosis are currently limited to investigational use but may be available at some medical centers.
Gigantism and Acromegaly
Background
Gigantism refers to abnormally high linear growth due to excessive action of insulin-like growth factor-I (IGF-I) while the epiphyseal growth plates are open during childhood. Acromegaly is the same disorder of IGF-I excess when it occurs after the growth plate cartilage fuses in adulthood. Gigantism is a nonspecific term that refers to any standing height more than 2 standard deviations above the mean for the person’s sex, age, and Tanner stage (ie, height Z score >+2). These disorders are placed along a spectrum of IGF-I hypersecretion, wherein the developmental stage when such excess originates determine the principal manifestations. The onset of IGF-I hypersecretion in childhood or late adolescence results in tall stature. This article focuses on IGF-I excess with an onset during childhood.
The most remarkable example of a person with gigantism was Robert Wadlow, called the Alton giant, who stood
Image shows the coauthor with a statue of Robert Wadlow, who was called the
More recently, scientific breakthroughs in the molecular, genetic, and hormonal basis of growth hormone (GH) excess have provided important insights into the pathogenesis, prognosis, and treatment of this exceedingly rare disease.
Pathophysiology
Causes of excess IGF-I action may be divided into 3 categories: (1) those originating from primary GH excess released from the pituitary; (2) those caused by increased GH-releasing hormone (GHRH) secretion or hypothalamic dysregulation; and (3) hypothetically, those related to the excessive production of IGF-binding protein, which prolongs the half-life of circulating IGF-I.
By far, most people with giantism have GH-secreting pituitary adenomas or hyperplasia. Although gigantism is typically an isolated disorder, rare cases occur as a feature of other conditions, such as multiple endocrine neoplasia (MEN) type I, McCune-Albright syndrome (MAS), neurofibromatosis, tuberous sclerosis, or Carney complex.
Approximately 20% of patients with gigantism have MAS (the triad of precocious puberty, café au lait spots, fibrous dysplasia) and may have either pituitary hyperplasia or adenomas
Epidemiology
Frequency
Gigantism is extremely rare, with approximately 100 reported cases to date. Acromegaly is more common than giantism, with an incidence of 3-4 cases per million people per year and a prevalence of 40-70 cases per million population.
Mortality/Morbidity
Because of the small number of people with gigantism, mortality and morbidity rates for this disease during childhood are unknown.
For individuals with acromegaly, the mortality rate is 2-3 times that of the general population. Successful treatment, with normalization of IGF-I levels, may be associated with a return to normal life expectancy. For persons with acromegaly, the most frequent causes of death are cardiovascular and respiratory complications. Transgenic mouse models of acromegaly demonstrate cardiac and vascular hypertrophy but normal function, raising the concern that hypertrophic cardiomyopathy may contribute to the increased mortality.
Researchers disagree on whether malignancy is a significant cause of increased mortality. Although benign tumors (including uterine myomas, prostatic hypertrophy, and skin tags) are frequently encountered in acromegaly, documentation for overall prevalence of malignancies in patients with acromegaly remains controversial. Most studies suggest that as many as 30% of patients may have a premalignant colon polyp at diagnosis, and as many as 5% may have a colonic malignancy. However, the long-term effect of colonic lesions on morbidity and mortality has not been established.
No clear evidence supports an increased risk for lung, breast, or prostate cancer. As a significant cause of morbidity, sleep apnea may be both obstructive and central.
Race
No predilection has been reported.
Sex
IGF-I excess equally affects men and women.
In a series of 12 children, GH-secreting adenomas occurred with a female-to-male ratio of 1:2. Given the small size of this series, these disorders are unlikely to show a sex bias during childhood.
Age
Gigantism may begin at any age before epiphyseal fusion. The mean age for onset of acromegaly is in the third decade of life. For acromegaly, the delay from the insidious onset of symptoms to diagnosis is 5-15 years, with a mean delay of 8.7 years.
Hyperaldosteronism
Background
Aldosterone is a steroid hormone produced exclusively in the zona glomerulosa of the adrenal cortex. It is the major circulating mineralocorticoid in humans. Numerous aldosterone precursors, including deoxycorticosterone and 18-hydroxycorticosterone, have mineralocorticoid activity and may produce or exacerbate features typical of mineralocorticoid hypertension when present in excessive amounts in various pathologic states.
The principal site of action of aldosterone is the distal nephron, though several other sites of aldosterone-sensitive sodium regulation are noted, including the sweat glands and the gastrointestinal (GI) tract. The principal regulators of aldosterone synthesis and secretion are the renin-angiotensin system and the potassium ion concentration. Minor regulators include adrenocorticotropic hormone (ACTH) from the pituitary, atrial natriuretic peptide from the heart, and local adrenal secretion of dopamine.
Hyperaldosteronism is characterized by excessive secretion of aldosterone, which causes increases in sodium reabsorption and loss of potassium and hydrogen ions. It may be either primary (autonomous) or secondary. Hyperaldosteronism represents part of a larger entity of hypermineralocorticoidism that may be caused by aldosterone, its mineralocorticoid precursors, or defects that modulate aldosterone effects on its target tissues.
Pathophysiology
Normal aldosterone physiology
Aldosterone participates in the homeostasis of circulating blood volume and serum potassium concentration; these, in turn, feed back to regulate aldosterone secretion by the zona glomerulosa of the adrenal cortex. Aldosterone secretion is stimulated by an actual or apparent depletion in blood volume detected by stretch receptors and by an increase in serum potassium ion concentrations; it is suppressed by hypervolemia and hypokalemia.
The mechanisms regulating aldosterone secretion are complex, involving the zona glomerulosa of the adrenal glands, the juxtaglomerular apparatus in the kidneys, the cardiovascular system, the autonomic nervous system, the lungs, and the liver (see the image below). The major factors stimulating aldosterone production and release by the zona glomerulosa are angiotensin II and the serum potassium concentration. The juxtaglomerular apparatus is the principal site of regulation of angiotensin II production.
ACTH stimulates aldosterone secretion in an acute and transient fashion but does not appear to play a significant role in the long-term regulation of mineralocorticoid secretion. The major inhibitors of the zona glomerulosa include circulating atrial natriuretic peptide (ANP) and, locally, dopamine. Although ANP levels are clearly increased in hyperaldosteronism, neither ANP nor dopamine has been implicated as a primary cause of clinically disordered aldosterone secretion.
Metoclopramide has been shown to increase aldosterone secretion, suggesting that dopamine may tonically inhibit aldosterone release. The physiologic roles of adrenomedullin and vasoactive intestinal peptide (VIP) on aldosterone secretion remain to be clarified, although both of these neuropeptides are produced in rat zona glomerulosa.
The synthesis of prorenin, its conversion to renin, and its systemic secretion are stimulated by blood volume contraction detected by stretch receptors, beta-adrenergic stimulation of the sympathetic nervous system, and prostaglandins I2 and E2. These processes are inhibited by volume expansion and ANP.
Renin converts angiotensinogen, a proenzyme synthesized in the liver, into the decapeptide angiotensin I, which is then converted in the lungs into the octapeptide angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II is both a stimulator of aldosterone secretion and a potent vasopressor. Angiotensin II is metabolized to angiotensin III, a heptapeptide that is also a stimulator of aldosterone secretion.
The synthesis and secretion of prostaglandins I2 and E2 and the normal function of the stretch receptors are dependent on the intracellular ionized calcium concentration. Renal prostaglandin secretion is stimulated by catecholamines and angiotensin II. The complex regulation of aldosterone synthesis and secretion provides several points at which disturbance in the regulation of aldosterone secretion may occur.
Aldosterone is synthesized from choleste rol in a series of 6 biosynthetic steps (see the image below). Only the las t 2 steps are specific to aldosterone synthesis; the first 4 also apply to cortisol synthesis by the zona fasciculata. Consequently, a defect in one of the specific aldosterone synthetic enzymes does not lead to hypercortisolism and secondary ACTH-mediated adrenal hyperplasia.
The enzyme aldosterone synthase is encoded by the gene CYP11B2 and has 11β-hydroxylase, 18-hydroxylase, and 18-hydroxydehydrogenase activity. This gene is located on human chromosome arm 8q24.3-tel, close to the gene CYP11B1, which encodes 11β-hydroxylase, the enzyme that catalyzes the final step of cortisol synthesis. Mutations in these genes can result in a number of disorders of aldosterone synthesis (see Differentials).
Aldosterone action on target tissues (eg, the distal renal tubule, sweat glands, salivary glands, and epithelium of the large intestine) is mediated via a specific mineralocorticoid receptor. Mineralocorticoid receptors exhibit equal affinity for mineralocorticoids and cortisol, yet the aldosterone receptors in the distal tubule and elsewhere are protected from cortisol-mediated activation by 11β-hydroxysteroid dehydrogenase type 2, which locally converts cortisol to inactive cortisone.
Primary aldosteronism
The term primary hyperaldosteronism (or primary aldosteronism) refers to a renin-independent increase in the secretion of aldosterone. This condition is principally a disease of adulthood, with its peak incidence in the fourth to sixth decades of life.
Approximately 99% of cases of primary aldosteronism are due either to an aldosterone-producing adenoma (APA), which accounts for approximately 40% of cases, or to idiopathic hyperaldosteronism (IHA), which accounts for approximately 60% of cases (almost all of which are bilateral). Adrenocortical carcinomas that are purely aldosterone-secreting are exceedingly rare and are usually large at the time of diagnosis. Unilateral adrenocortical hyperplasia is a rare occurrence.
APAs (sometimes referred to as aldosteronomas) are usually benign encapsulated adenomas that are less than
Patients with IHA have bilateral thickening and variable nodularity of their adrenal cortex. A wide spectrum of severity exists for this disorder, which may go undetected for long periods with no hypokalemia and only mild hypertension. It has been suggested that IHA arises as a result of an undetected adrenal cortex–stimulating factor. Alternatively, the disorder may arise as a result of an activating mutation in an adrenal cortex–specific gene. Neither hypothesis has been proven.
Inherited forms of primary hyperaldosteronism account for only 1% of cases but are more likely to occur during childhood years. These forms include familial hyperaldosteronism (FH) types I and II.
Familial hyperaldosteronism type I
FH type I (FH-I), also referred to as glucocorticoid-remediable aldosteronism (GRA), may be detected in asymptomatic individuals during screening of the offspring of affected individuals, or patients may present in infancy with hypertension, weakness, and failure to thrive due to hypokalemia. FH-I is inherited in an autosomal dominant manner and has a low frequency of new mutations.
The first clinical description of GRA appeared in 1966, and the genetic mechanism was discovered in 1992. FH-I arises as a result of unequal crossing over of highly related CYP11B1 (the 11β-hydroxylase gene) and CYP11B2 (the aldosterone synthase gene) during meiosis, producing an anti-Lepore-type fusion product.This genetic rearrangement causes the expression of CYP11B2 to be placed under the control of the CYP11B1 promoter and the aldosterone synthesis to be abnormally regulated by ACTH rather than by the renin-angiotensin system.
The result is ACTH-dependent aldosterone production and production of 17-hydroxylated analogues of 18-hydroxycortisol under ACTH regulation from ectopic enzyme expression in the zona fasciculata. Bilateral hyperplasia of the zona fasciculata occurs, and high levels of novel 18-hydroxysteroids appear in the urine. Adenoma formation is rare, but patients do have a significant increase in incidence of cerebrovascular aneurysms, for which they require screening.
Familial hyperaldosteronism type II
FH type II (FH-II) is a non–glucocorticoid-suppressible inherited form of hyperaldosteronism that was first recognized as a distinct entity by Gordon et al, though cases had previously been described in the 1980s. Like FH-I, it is inherited in an autosomal dominant manner. In contrast to FH-I, some FH-II kindreds exhibit a high rate of adenoma formation.
The mechanism and gene locus have not yet been identified, though CYP11B and the renin and angiotensin II receptor genes have been excluded. However, linkage has been established for a number of families to band 7p22.It has also been speculated that FH-II is not a single disorder.
Familial hyperaldosteronism type III
FH-III is a rare autosomal dominant form of PA characterized by early-onset hypertension, nonglucocorticoid-remediable hyperaldosteronism, and hypokalemia. Germline heterozygous missense mutations of the KCNJ5 gene, encoding Kir3.4, a member of the inwardly rectifying K channel family, have been identified as a cause of FH-III. Thus far, 4 mutations (G151R, G151E, T158A, and I157S) have been reported in 6 families.
The clinical phenotype of patients harboring the above mutations ranges from severe primary aldosteronism and hypertension refractory to medical treatment that requires bilateral adrenalectomy, to mild or moderate hypertension responsive to medical therapy. In some patients, adrenal hyperplasia has been described.
Various studies from different centers report a prevalence of somatic KCNJ5 mutations in sporadic APAs ranging from 30-65%.There are 2 recurrent mutations, G151R and L168R, reported by all studies, whereas there is one report of a 3-nucleotide deletion, the delI157.
The affected residues of both the germline and the somatic mutations are in or near the selectivity filter of the Kir3.4 potassium channel and are highly conserved among different species. Electrophysiologic studies demonstrate that these mutations result in loss of channel selectivity, with increased Na conductance leading to membrane depolarization. In zona glomerulosa cells, membrane depolarization leads to opening of voltage activated Ca channels, with activation of the calcium-signalling pathway, the major mediator of aldosterone production.
APAs with KCNJ5 mutations are more prevalent in females than males and in younger patients. They are also associated with higher preoperative aldosterone levels. They are not related with the tumor size, but they are related with higher aldosterone levels and lower K concentrations.
Transcriptome and real-time polymerase chain reaction (PCR) analyses demonstrate that APAs with KCNJ5 mutations exhibit increased expression of the CYP11B2 gene and its transcriptional regulator NR4A2, thus increasing aldosterone production. It has also been found that APAs with and without KCNJ5 mutations display slightly different gene expression patterns.Another study reports KCNJ5 mRNA levels higher in the APAs with KCNJ5 mutations and significantly higher in APA than cortisol-producing adenomas and pheochromocytomas.
Secondary hyperaldosteronism
Secondary hyperaldosteronism is a collective term for a diverse group of disorders characterized by physiologic activation of the renin-angiotensin-aldosterone (R-A-A) axis as a homeostatic mechanism designed to maintain serum electrolyte concentrations or fluid volume. In the presence of normal renal function, it may lead to hypokalemia.
Secondary hyperaldosteronism can be divided into 2 categories, 1 with associated hypertension and 1 without. The former category includes renovascular hypertension, which results from renal ischemia and hypoperfusion leading to activation of the R-A-A axis. The most common causes of renal artery stenosis in children are fibromuscular hyperplasia and neurofibromatosis. Hypokalemia may occur in as many as 20% of patients.
Plasma renin activity (PRA) levels are often in the reference range, but elevated levels of PRA may be detected after provocation with a single dose of captopril 1 mg/kg. Renal ischemia is also thought to underlie the secondary hyperaldosteronism observed in malignant hypertension.
Hyperreninemia and secondary aldosteronism have also been reported in patients with pheochromocytoma, apparently as a result of functional renal artery stenosis. Renin-producing tumors are very rare, and very high levels of PRA (up to 50 ng/mL/h) are noted, frequently with an increased prorenin-to-renin ratio. The tumors are generally of renal origin and include Wilms tumors and renal cell carcinomas.
Hyperkalemia due to chronic renal failure also causes secondary hyperaldosteronism. Low sodium-to-potassium ratios can be measured in saliva and stool. Cyclosporine-induced hypertension in solid organ transplant patients may also involve a component of hyperaldosteronism.
Secondary hyperaldosteronism in the absence of hypertension occurs as a result of homeostatic attempts to maintain the sodium concentration or circulatory volume or to reduce the potassium concentration. Clinical conditions in which it may arise include diarrhea, excessive sweating, low cardiac output states, and hypoalbuminemia due to liver or renal disease or nephrotic syndrome. Secondary hyperaldosteronism may also occur developmentally iewborn infants (see below).
Increased mineralocorticoid dependency in the young
The mineralocorticoid dependency of sodium reabsorption is increased during infancy and childhood, peaking in the neonatal period before decreasing progressively with advancing age. This increase occurs because the reabsorption of sodium and water by the proximal tubule is least efficient in early life, resulting in an increased sodium and water load at the level of the distal renal tubule.
Because sodium and water resorption from the distal tubule is mediated by the R-A-A axis, the PRA is approximately 10-fold to 20-fold higher in a newborn infant than in an adult. Consequently, neonates show relative increases in aldosterone production rates (>300 µg/m/day vs 50 µg/m/day in an adult) and plasma aldosterone concentrations (80 pg/dL vs 16 pg/dL). These increases in early life explain why young infants exhibit profound clinical symptoms of hyperaldosteronism that gradually improve with advancing age.
Etiology
The following is a summary of etiologies of hyperaldosteronism and conditions that mimic hyperaldosteronism:
Causes of primary hyperaldosteronism include the following:
- APA – High aldosterone, low PRA
- IHA – Responds to posture (bilateral adrenal hyperplasia)
- Primary adrenal hyperplasia – Responds to posture (unilateral disease)
- FH-I (GRA) – Sustained suppression of aldosterone (< 4 ng/dL) with dexamethasone
- FH-II/FH-III – Familial (probably autosomal dominant)
Causes of secondary hyperaldosteronism include the following:
- Edema disorders (eg, cardiac failure and nephrotic syndrome) – High aldosterone, nonsuppressed PRA (>2 ng/mL)
- Renovascular hypertension
- Renin-producing tumors
- Pregnancy
Causes of conditions that mimic aldosterone excess include the following:
- Congenital adrenal hyperplasia (11β-hydroxylase deficiency and 17α-hydroxlyase deficiency) – Low aldosterone, low PRA, elevated steroid intermediates
- Primary glucocorticoid resistance – High glucocorticoid secretion unsuppressed by dexamethasone
- Deoxycorticosterone-secreting tumors – Elevated deoxycorticosterone levels
- Syndrome of apparent mineralocorticoid excess
- Liddle syndrome
- Licorice ingestion
- Carbenoxolone
Hypokalemia may be precipitated by a diet that is rich in sodium or the concomitant administration of drugs that produce kaliuresis (including diuretics and carbenoxolone). Taking carbenoxolone or eating large quantities of licorice may result in hypokalemia because of blockade of the target tissue enzyme that protects the aldosterone receptor from the relatively higher levels of circulating cortisol (apparent mineralocorticoid excess).
Epidemiology
Primary hyperaldosteronism is a rare condition in children. The youngest child reported with an aldosterone-secreting adenoma was aged 3 years. Earlier use of hypokalemia as a diagnostic requirement, as advocated by some authorities, may have led to underrecognition of the contribution of primary hyperaldosteronism to hypertension.
A study that used saline infusion as a screening test for primary aldosteronism reported a frequency of 2.2% of primary hyperaldosteronism among 1036 unselected adults with hypertension.A smaller study that used the aldosterone-to-PRA ratio in plasma suggested that primary hyperaldosteronism might account for an even greater proportion of cases of hypertension.
Most of the hyperaldosteronism observed in the general population is sporadic, with most cases due to bilateral adrenal hyperplasia. APAs are likely to be diagnosed earlier than IHA because they are more likely than IHA to produce early symptomatic hypertension and hypokalemia. APAs account for 40% of cases of primary hyperaldosteronism.
It is possible that the distinction between adenoma and hyperplasia is not as clear as was once assumed. In one third of cases, associated hyperplasia or nodules of the adjacent zona glomerulosa is present, implying that the adenoma may have arisen in previously hyperplastic tissue.
Inherited forms of primary hyperaldosteronism (ie, FH-I [GRA], FH-II, and a very rare form known as FH type III [FH-III]) account for approximately 1% of cases of primary hyperaldosteronism, though they are more likely to occur during childhood and adolescent years than other forms of primary hyperaldosteronism are.
Studies of secondary hyperaldosteronism have found that approximately 15% of adults who attend hypertension clinics have elevated PRA. Reliable figures for children are not readily available.
Age-, sex-, and race-related demographics
Because the 2 causes that account for about 99% of cases of primary hyperaldosteronism have a peak age of onset in adulthood, the less common causes account for a larger percentage of children with hyperaldosteronism. For this reason, children with apparent hyperaldosteronism should be evaluated for evidence of congenital defects of the R-A-A axis and inherited forms of hypermineralocorticoidism.
Data on adults suggest that hyperaldosteronism has a female preponderance. Equivalent information is not available for children, in whom primary hyperaldosteronism due to inherited syndromes is likely to represent a greater proportion of cases.
The literature on adults demonstrates that blacks are at significantly greater risk for hypertension-related morbidity and mortality than whites are. They are also more likely to develop low-renin hypertension, though no studies indicate that the prevalence of primary hyperaldosteronism is significantly higher in blacks.
Prognosis
The age of the patient and the duration of disease before diagnosis are the 2 most important prognostic factors. Adult studies have shown that hypertension is improved significantly in approximately 70% of cases (see Treatment). This figure is likely to be higher in children because disease duration is shorter and the prevalence of other causes of hypertension is lower.
Primary hyperaldosteronism can result in substantial morbidity and mortality as a result of hypertensive vascular complications (hypertrophy followed by sclerosis of intimal smooth muscle), renal complications (sclerosis), and cardiac complications (hypertrophy followed by dilatation). Through early recognition and treatment of hypertension, these complications can be avoided in children.
Patients with GRA must undergo assessment of their cerebral circulation because this disorder is associated with a significant risk of cerebral vascular aneurysms. Provided that hypertension is well treated, morbidity and mortality should not be increased significantly.
Hypokalemia is more frequently observed in patients with adenomas, though it should not be considered a diagnostic feature of primary hyperaldosteronism, as was once thought. Patients with adenomas are more likely to develop this complication, as are patients who have milder disease but receive treatment with diuretics for their hypertension before the hyperaldosteronism is diagnosed.
Hypokalemic patients may experience neuromuscular symptoms such as weakness or paralysis, constipation, and polyuria and polydipsia because of an associated renal concentrating defect. Hypokalemia also impairs insulin secretion and can promote the development of diabetes mellitus.
Although cardiac fibrosis has been reported in adults with primary hyperaldosteronism, no such reports exist in children, possibly because of the shorter duration of disease at the time of diagnosis. Cardiac fibrosis has also been reported in rats treated with excessive amounts of mineralocorticoids, especially if hyperglycemia is also present. This effect can be ameliorated with amiloride. The role of aldosterone in diabetic heart disease has been questioned, and trials of mineralocorticoid antagonists in this condition have been initiated.
Hyperpituitarism
Background
Hyperpituitarism, or primary hypersecretion of pituitary hormones, is rare in children. It typically results from a pituitary microadenoma. The most frequently encountered adenoma in children is the prolactinoma, followed by corticotropinoma and somatotropinoma. Fewer than 20 cases of thyrotropinoma in children have been reported, all with onset after age 11 years. Pediatric gonadotropinoma has not been reported.
Hypersecretion of pituitary hormones secondary to macroadenomas (see the image below) can interfere with other pituitary hormone functions, resulting in target organ hormone deficiencies (hypogonadism, hypoadrenalism, hypothyroidism).
In some cases, long-standing hormonal hypersecretion is accompanied by sufficient hyperplasia of the pituitary to produce sellar enlargement.
Elevated pituitary hormone levels that result from primary endocrine organ deficiency (eg, high circulating thyroid-stimulating hormone [TSH] levels in primary hypothyroidism due to Hashimoto thyroiditis) quickly suppress to reference range values upon replacement of the active hormone. Most rarely, ectopic tumors can secrete pituitary hormones. This article focuses on the endocrine manifestations of pituitary adenomas in children.
Pathophysiology
Hypothalamic dysfunction clearly may promote tumor growth, but overwhelming evidence indicates intrinsic pituicyte genetic disruption leads to pituitary tumorigenesis. The monoclonal nature of most pituitary adenomas, confirmed by X-inactivation studies, implies their usual origin from a clonal event in a single cell. Most pituitary adenomas are functional and secrete a hormone that produces a characteristic clinical presentation. Nonfunctioning pituitary adenomas are rare in children, accounting for only 3-6% of all adenomas in 2 large series, whereas they comprise 30% of adenomas in adults. In children, disruption of growth regulation and/or sexual maturation is common, either because of hormone hypersecretion or because of manifestations caused by local compression by the tumor.
Prolactinoma
Overall, prolactinoma is the most common pituitary adenoma encountered in childhood. Most pediatric cases occur in adolescence, more commonly in females than males. Boys tend to have larger tumors and higher serum prolactin (PRL) levels than girls. Females with these tumors present with amenorrhea, and males present with gynecomastia and hypogonadism. Prolactinomas arise from acidophilic cells that are derived from the same lineage as the somatotropes and thyrotropes. Hence, PRL-secreting adenomas may also stain for and secrete growth hormone (GH) and, occasionally, TSH.
Corticotropinoma (Cushing disease)
In children, corticotropinomas are the most common adenomas observed before puberty, although they occur in people of all ages. They increase in frequency in pubescent and postpubescent children, with a female preponderance. First described by Harvey Cushing in the early 1900s, Cushing disease (see the images below) specifically refers to an adrenocorticotropic hormone (ACTH)–producing pituitary adenoma that stimulates excess cortisol secretion.
Adenomas that cause Cushing disease are significantly smaller than all other types of adenomas at presentation. Children have clinical courses somewhat different from adults. They most commonly present with weight gain (usually not centripetal) and growth failure. As in adults, most patients display an absence of the physiologic diurnal rhythm of plasma cortisol and ACTH with increased urinary excretion of free cortisol and 17-hydroxycorticosteroids (17-OHCS).
Somatotropinoma (gigantism)
GH-secreting adenomas are rare in childhood. Gigantism refers to GH excess in childhood when open epiphysial plates allow for excessive longitudinal growth. Most cases of gigantism result from GH-secreting pituitary adenomas or hyperplasia. Although gigantism typically occurs as an isolated disorder, it occasionally represents one feature of other conditions (eg, multiple endocrine neoplasia [MEN] type 1, McCune-Albright syndrome [MAS], neurofibromatosis, tuberous sclerosis, Carney complex).
Mammosomatotrophs are the most common type of GH-secreting cells in childhood gigantism; hence, GH-secreting adenomas often stain for and secrete PRL (67% in one study). GH-secreting tumors in pediatric patients are more likely to be locally invasive or aggressive than those in adult patients. Activating mutations of the stimulatory Gs alpha (Gsa) protein have been identified in the somatotrophs of pituitary lesions in MAS and in as many as 40% of sporadic GH-secreting pituitary adenomas.
Thyrotropinoma
Very few cases of thyrotropinoma have been reported in children. These adenomas may secrete excess PRL, GH, and alpha subunit in addition to TSH. They are usually large because of their aggressive features and because their diagnosis is often delayed. The clinical presentation consists of signs and symptoms of hyperthyroidism, visual symptoms, and headaches. Biochemical features include the elevation of circulating free thyroxine (T4) and total triiodothyronine (T3) levels but inappropriately unsuppressed TSH.
Epidemiology
Frequency
Although less common in children than in adults, pituitary adenomas constitute 2.7% of supratentorial tumors in children and 3.6-6% of all pituitary adenomas that are surgically treated. The average annual incidence of pituitary adenomas presenting before age 20 years is estimated to be less than 0.1 per million children.
Mortality/Morbidity
Transsphenoidal pituitary surgery has emerged as the treatment of choice for ACTH-secreting and GH-secreting adenomas. Transsphenoidal surgery is indicated for prolactinomas that do not respond to medical therapy. Transsphenoidal surgery is associated with remarkably little morbidity and near zero mortality. A permanent loss of pituitary function occurs infrequently. The incidence of postoperative hypopituitarism is about 3% in patients with microadenomas and slightly increases with the invasiveness of the tumor.
Race
Race and ethnicity have not been reported as significant contributing factors to hyperpituitarism.
Sex
In prolactinoma, the female-to-male ratio is 4.5:1. In ACTH-releasing adenoma, the female-to-male ratio is 2:1. In GH-releasing adenoma, the female-to-male ratio is 1:2.
Age
In children, ACTH-releasing adenomas are most prevalent in the youngest group and decrease in frequency with advancing age.
The incidence of prolactinomas increases with age; 93% occur in children older than 12 years.
GH-releasing tumors have a fairly even distribution among the various age groups
Hypogonadism
Practice Essentials
Morbidity for men and women with hypogonadism includes infertility and an increased risk of osteoporosis; there is no increase in mortality.
Signs and symptoms
History
Considerations in the evaluation of males with hypogonadism include the following:
- Developmental anomalies associated with the genital system (eg, hypospadias, micropenis, and cryptorchidism)
- For postpubertal males, the rate of beard growth, libido and sexual function, muscle strength, and energy levels
- Possible causes of acquired testicular failure (eg, mumps orchitis, trauma, radiation exposure of the head or testes, and chemotherapy)
- Drugs that may interrupt testicular function: Including agents that interfere with testosterone synthesis, such as spironolactone, cyproterone, marijuana, heroin, and methadone
Considerations in the evaluation of females with hypogonadism include the following:
- Signs associated with Turner syndrome (eg, lymphedema, cardiac or renal congenital anomalies, and short growth pattern)
- Age of menarche
Physical examination
Considerations in the physical examination of males with hypogonadism include the following:
- Evaluation of the testes: This is the most important feature of the physical examination; determine whether both testes are palpable, their position in the scrotum, and their consistency; testes size can be quantitated by comparison with testicular models (orchidometer), or their length and width may be measured
- Examination of the genitalia for hypospadias
- Examination of the scrotum to see if it is completely fused
- Evaluation of the extent of virilization
- Staging of puberty: Use the Tanner criteria for genitalia, pubic hair, and axillary hair
- Examination for signs of Klinefelter syndrome (eg, tall stature, especially if the legs are disproportionately long, gynecomastia, small or soft testes, and a eunuchoid body habitus)
Considerations in the physical examination of females with hypogonadism include the following:
- Examination of the genitalia is important
- Determination of the extent of androgenization: May be adrenal or ovarian in origin and is demonstrated in pubic and axillary hair
- Determination of the extent of estrogenization: As evidenced by breast development and maturation of the vaginal mucosa
- Examination for signs of Turner syndrome (eg, short stature, webbing of the neck [such as pterygium colli], a highly arched palate, short fourth metacarpals, widely spaced nipples, or multiple pigmented nevi)
Diagnosis
The following studies may be indicated in males with hypogonadism:
- Follicle-stimulating hormone (FSH) levels
- Luteinizing hormone (LH) levels
- Prolactin levels
- Testosterone levels
- Thyroid function
- Seminal fluid examination
- Karyotyping
- Testicular biopsy
For males after puberty, the Guidelines of the Endocrine Societyrequire that the diagnosis of hypogonadism be based on symptoms and signs of hypogonadism plus the presence of a low testosterone level measured on at least 2 occasions.
The following studies may be indicated in females with hypogonadism:
- FSH levels
- LH levels
- Prolactin levels
- Estradiol levels
- Antiovarian antibody levels: If gonadotropin levels are elevated
- Thyroid function
- Karyotyping
Additional tests in the evaluation of patients with hypogonadism include the following:
- Adrenocorticotropic hormone (ACTH) stimulation testing: In patients in whom a form of congenital adrenal hyperplasia is suspected, adrenal steroid synthesis is best evaluated by performing a cosyntropin (ACTH 1-24) stimulation test
- Luteinizing-hormone releasing hormone (LHRH) stimulation testing: To distinguish between true hypogonadotropic hypogonadism and constitutional delay in growth and maturation
- Testicular tissue testing: If the testes are not palpable and if it is not certain whether any testicular tissue is present, administering human chorionic gonadotropin (hCG) and measuring testosterone response may be helpful
Management
Hormonal replacement
The simplest and most successful treatment for males and females with either hypergonadotropic or hypogonadotropic hypogonadism is replacement of sex steroids, but the therapy does not confer fertility or, in men, stimulate testicular growth.
When fertility is desired, an alternative therapy for men with hypogonadotropic hypogonadism is administration of pulsatile LHRH or injections of hCG and FSH. (In patients with hypergonadotropic hypogonadism, fertility is not possible.)
In a 6-year European study of men being treated for hypogonadism, long-term transdermal testosterone treatment did not increase prostate-specific antigen (PSA) levels or influence prostate cancer risk.
Investigators used data from a 5-year, open-label extension of a 1-year trial of a transdermal testosterone patch (Testopatch) in men with hypogonadism. Study subjects wore two
Background
Hypogonadism manifests differently in males and in females before and after the onset of puberty.If onset is in prepubertal males and testosterone replacement is not instituted, the individual has features of eunuchoidism, which include sparse body hair, poor development of skeletal muscles, and delay in epiphyseal closure, resulting in long arms and legs. When hypogonadism occurs in postpubertal males, lack of energy and decreased sexual function are the usual concerns. In females with hypogonadism before puberty, failure to progress through puberty or primary amenorrhea is the most common presenting feature. When hypogonadism occurs in postpubertal females, secondary amenorrhea is the usual concern.
Pathophysiology
The gonad (ovary or testis) functions as part of the hypothalamic-pituitary-gonadal axis. A hypothalamic pulse generator resides in the arcuate nucleus, which releases luteinizing hormone (LH)-releasing hormone (LHRH), which is also termed gonadotropin-releasing hormone (GnRH), into the hypothalamic-pituitary portal system. Data suggest that a gene named KISS is important in the development of the LHRH-secreting cells.
In response to these pulses of LHRH, the anterior pituitary secretes follicle-stimulating hormone (FSH) and LH, which, in turn, stimulate gonadal activity. The increase in gonadal hormones results in lowered FSH and LH secretion at the pituitary level, completing the feedback loop. In the testes, LH stimulates Leydig cells to secrete testosterone, whereas FSH is necessary for tubular growth. In the ovaries, LH acts on theca and interstitial cells to produce progestins and androgens, and FSH acts on granulosa cells to stimulate aromatization of these precursor steroids to estrogen.
Hypogonadism may occur if the hypothalamic-pituitary-gonadal axis is interrupted at any level. Hypergonadotropic hypogonadism (primary hypogonadism) results if the gonad does not produce the amount of sex steroid sufficient to suppress secretion of LH and FSH at normal levels. Hypogonadotropic hypogonadism may result from failure of the hypothalamic LHRH pulse generator or from inability of the pituitary to respond with secretion of LH and FSH. Hypogonadotropic hypogonadism is most commonly observed as one aspect of multiple pituitary hormone deficiencies resulting from malformations (eg, septooptic dysplasia, other midline defects) or lesions of the pituitary that are acquired postnatally. In 1944, Kallmann and colleagues first described familial isolated gonadotropin deficiency. Recently, many other genetic causes for hypogonadotropic hypogonadism have been identified.
Normosmic hypogonadotropic hypogonadism, in which the sense of smell is not disrupted, has been associated with mutations in GNRH1, KISS1R, and GNRHR genes. Although their exact functions are unclear, the genes TAC3 and TACR3 have also been associated with normosmic hypogonadotropic hypogonadism. Kallmann syndrome (anosmic hypogonadotropic hypogonadism) has been associated with mutations in KAL1, FGFR1, FGF8, PROK2, and PROKR2 genes. The relationship with Kallmann syndrome is thought to be because these genes are all related to the development and migration of GnRH neurons. Mutations of an additional gene, CHD7, which has been associated with CHARGE syndrome, has also been found in patients with both normosmic or anosmic hypogonadotropic hypogonadism.
Frequency
International
In women with hypergonadotropic hypogonadism (ie, gonadal failure), the most common cause of hypogonadism is Turner syndrome, which has an incidence of 1 case per 2,500-10,000 live births. In men with hypergonadotropic hypogonadism, the most common cause is Klinefelter syndrome, which has an incidence of 1 case per 500-1000 live births. Hypogonadotropic hypogonadism is more rare.
Mortality/Morbidity
No increase in mortality is observed in patients with hypogonadism. Morbidity for men and women includes infertility and an increased risk of osteoporosis. In women, an increased risk of severe osteoporosis is noted. In men, hypogonadism causes decreased muscle strength and sexual dysfunction.
Race
No racial predilection has been described.
Sex
Hypergonadotropic hypogonadism is more common in males than in females because the incidence of Klinefelter syndrome (the most common cause of primary hypogonadism in males) is higher than the incidence of Turner syndrome (the most common cause of hypogonadism in females). Incidence of hypogonadotropic hypogonadism is equal in males and females.
Age
Hypogonadism may occur at any age; however, consequences differ according to the age at onset. If hypogonadism occurs prenatally (even if incomplete), sexual ambiguity may result. If hypogonadism occurs before puberty, puberty does not progress. If hypogonadism occurs after puberty, infertility and sexual dysfunction result.
Panhypopituitarism
Background
The pituitary gland is called the master endocrine gland of the body because it controls the function of other endocrine organs. The anterior pituitary produces the hormones thyrotropin (thyroid-stimulating hormone [TSH]), corticotropin (adrenocorticotropic hormone [ACTH]), luteinizing hormone (LH), follicle-stimulating hormone (FSH), growth hormone (GH), and prolactin (PRL). The anterior pituitary is controlled by specific hypothalamic-releasing hormones. The posterior pituitary produces vasopressin (antidiuretic hormone [ADH]) and oxytocin.
Panhypopituitarism is a condition of inadequate or absent production of the anterior pituitary hormones. It is frequently the result of other problems that affect the pituitary gland and either reduce or destroy its function or interfere with hypothalamic secretion of the varying pituitary-releasing hormones. Panhypopituitarism can be the end result of various clinical scenarios. The signs and symptoms are diverse. Manifestations of congenital anterior hypopituitarism include micropenis, midline defects, optic atrophy, hypoglycemia, and poor growth.
Pathophysiology
The effects of hypopituitarism in children depend on the affected hormones. GH deficiency can result in hypoglycemia and short stature. Gonadotropin deficiency leads to prenatal micropenis and delayed or interrupted puberty in older children. Corticotropin deficiency interferes with normal carbohydrate, protein, and lipid metabolism and may result in weight loss, hypoglycemia, fatigue, hypotension, and death. Thyrotropin deficiency leads to hypothyroidism.
Epidemiology
Frequency
Hypopituitarism is caused by various conditions and is associated with various hormonal deficiencies. Thus, data are limited regarding frequency rates of the various etiologies and components. An Italian study reported GH deficiency prevalence to be approximately 9 cases per 1000 individuals in a pediatric population.Data from the Northwest Regional Screening program estimate the frequency of congenital TSH deficiency at 1 case per 29,000 live births.
Mortality/Morbidity
Morbidity and mortality due to hypopituitarism are caused by the individual hormone deficiencies or the underlying cause of hypopituitarism. Individual hormonal deficiencies are discussed in greater detail in the specific articles, and the underlying causes of death are not discussed here.
Acute mortality due to hormonal deficiencies is rare. When deaths occur due to hormonal deficiencies, they are usually caused by adrenal insufficiency secondary to ACTH deficiency. These deaths are most likely to occur when an accompanying illness prevents appropriate oral glucocorticoid replacement.
Growing, but not completely conclusive, evidence indicates that childhood hypopituitarism may be associated with a shortened adult lifespan, even with adequate hormonal replacement.The increased mortality is due to cardiovascular abnormalities that are related to GH deficiency and past practices of not treating a GH deficiency when growth is complete.Children and adolescents with GH deficiency have been shown to have impaired vascular function.In adults, GH treatment restores improves cardiovascular risk factors, but long-term studies that demonstrate reduced cardiovascular disease have not been reported.
Pediatric Adrenal Insufficiency (
Background
Adrenal insufficiency (
Adrenal insufficiency (
Anatomy
The adrenal cortex is divided into 3 major anatomic zones. The zona glomerulosa produces aldosterone, and the zonae fasciculata and reticularis together produce cortisol and adrenal androgens. A fetal zone, unique to primates, produces dehydroepiandrosterone (DHEA), a precursor of both androgens and estrogens. This zone involutes within the first few months of postnatal life.
Aldosterone secretion is primarily regulated by the renin-angiotensin system. Increased serum potassium concentrations can also stimulate aldosterone secretion. Cortisol secretion is regulated by adrenocorticotropic hormone (ACTH), which, in turn, is regulated by corticotropin-releasing hormone (CRH) from the hypothalamus. Serum cortisol inhibits the secretion of both CRH and ACTH to prevent excessive secretion of cortisol from the adrenal glands.
ACTH partially regulates adrenal androgen secretion; other unknown factors contribute to this regulation as well. ACTH not only stimulates cortisol secretion but also promotes growth of the adrenal cortex in conjunction with growth factors such as insulinlike growth factor (IGF)-1 and IGF-2.
Etiology
Iatrogenic central adrenal insufficiency as well as acquired and congenital primary adrenal insufficiency (Addison disease) are briefly discussed in this section.
Iatrogenic central adrenal insufficiency
Most cases of adrenal insufficiency (
Treatment with megestrol acetate, an orexigenic agent, has also resulted in iatrogenic adrenal suppression. The mechanism is presumably related to the glucocorticoid properties of megestrol acetate.
Other causes of central adrenal insufficiency include congenital or acquired hypopituitarism and ACTH unresponsiveness. This unresponsiveness may be isolated (as in Familial Glucocorticoid Deficiency) (Online Mendelian Inheritance in Man database [OMIM] 202200),or it may be associated with achalasia and alacrima (as in achalasia-addisonism-alacrima syndrome, or triple A syndrome [AAAS]) (OMIM 231550).
Acquired primary adrenal insufficiency
In developed countries, the most common cause of adrenal insufficiency (
Patients with type 1 PGAD (OMIM 240300) usually present in the first decade of life with mucocutaneous candidiasis or hypoparathyroidism. This is an autosomal recessive disorder that involves the AIRE gene on chromosome 21 and presents with all or some of the following features:
- Chronic mucocutaneous candidiasis
- Hypoparathyroidism
- Adrenal failure
- Gonadal failure
- Vitiligo
- Alopecia
- Hypothyroidism
- Type 1 diabetes mellitus
- Pernicious anemia
- Steatorrhea
Type 2 PGAD (Schmidt syndrome; OMIM 269200) consists of type 1 diabetes mellitus, autoimmune thyroid disease, and adrenal failure. Individuals with this condition generally present in the second or third decades of life, although some components of the syndrome may be present in the pediatric age group. Type 2 PGAD is transmitted as an autosomal disorder with variable penetrance.
Other acquired causes of adrenal failure include the following:
- Adrenal hemorrhage
- Infections (eg, tuberculosis [TB], human immunodeficiency virus [HIV] infection)
- Neoplastic destruction
- Metabolic disorders (eg, various forms of adrenal leukodystrophy (OMIM 300100),Wolman disease [OMIM 278000], Smith-Lemli-Opitz syndrome (OMIM 270400))
- Administration of the anesthetic agent etomidate
Hemochromatosis may cause either primary (hereditary form OMIM 235200) or secondary adrenal insufficiency. Among patients with thalassemia or other forms of anemia who have received multiple transfusions, iron deposition in the pituitary and/or adrenal glands may also cause adrenal insufficiency.
Congenital primary adrenal insufficiency
Congenital
Inherited as an X-linked disorder, adrenal hypoplasia congenita (OMIM 300200) is caused by deletion or mutation of the DAX1/NR0B1 gene on chromosome Xp21.2, and is additionally associated with hypogonadotrophic hypogonadism and primary defects in sperm production.This is often part of a contiguous gene deletion that also involves the genes for glycerol kinase deficiency and dystrophin, resulting in elevations in serum glycerol (often measured using a triglyceride assay) and Duchenne muscular dystrophy. An alternate form, non-X linked, is characterized by intrauterine growth retardation and skeletal and genital anomalies (ie, IMAGe syndrome) (OMIM 300290). A third type of familial of adrenal hypoplasia congenita of uncertain etiology has been described (OMIM 240200).
Congenital adrenal hyperplasia results from a deficiency of one of several enzymes required for adrenal synthesis of cortisol. Symptoms of adrenal insufficiency (
Lipoid adrenal hyperplasia is another rare form of adrenal insufficiency (
Mutations or deletions involving CYP oxidoreductase, a flavoprotein that provides electrons to various enzyme systems, results in combined deficiencies of 17-hydroxylase, 21-hydroxylase, and 17-20 lyase activities. The result is adrenal insufficiency (
Relative adrenal insufficiency
The term relative adrenal insufficiency (
Some patients developed adrenal insufficiency (
Among critically ill children, a low incremental cortisol response to ACTH does not predict mortality.There is still much controversy regarding how to best diagnose adrenal insufficiency in hospitalized children and adults, as well as whether and when to treat. Thus, the decision to treat a critically ill patient with glucocorticoids must be made on a case-by-case basis until further definitive evidence is available.
Epidemiology
Primary adrenal insufficiency (Addison disease) is uncommon in the
Willis and Vince collected data from
Worldwide, the most common cause of adrenal insufficiency (
Although there does not appear to be a racial predilection, sex and age-related differences have been observed. Autoimmune adrenal insufficiency (
A form of congenital adrenal hypoplasia due to a defect in DAX1/NR0B1 is also X-linked and, therefore, is confined to males. Secondary forms of adrenal insufficiency (
Congenital causes, such as congenital adrenal hyperplasia, congenital adrenal hypoplasia, and defects in the ACTH receptor, most commonly become apparent in childhood.
Prognosis
With proper treatment and compliance, patients with adrenal insufficiency (
Death is a common outcome—usually from hypotension or cardiac arrhythmia secondary to hyperkalemia—unless replacement steroid therapy is begun.
Complications
Hypotension, shock, hypoglycemia, and death are the primary complications of adrenal insufficiency (
- Growth failure
- Obesity
- Striae
- Osteoporosis
- Muscle weakness
- Hypertension
- Hyperglycemia
- Cataracts
Complications of excessive administration of mineralocorticoids include hypertension and hypokalemia.
Patient Education
Educate patients with adrenal insufficiency (
Advise patients and their caretakers to immediately seek medical help if the patient becomes ill. Patients should wear or carry a medical alert tag or card at all times to help them receive appropriate emergency care if they are found unconscious.
Supplemental and injectable glucocorticoid
Patients and their caretakers should know how to administer supplemental glucocorticoid in times of illness or traumatic stress. Include education about how to administer an injectable glucocorticoid when the patient is vomiting or unable to take oral stress doses. Periodically reinforce this information, because caretakers are often reluctant to inject medications.
An intramuscular injection of hydrocortisone (eg, 25 mg for infants, 50 mg for children, 100 mg for adults) can be lifesaving in the interval before the patient receives professional medical care. If this injection is not possible, rectal hydrocortisone can be used until systemic glucocorticoids can be administered.
Dietary considerations
Patients should eat an unrestricted diet. Those with primary adrenal insufficiency (
References
1. Nykytyuk S.O.,Balatska N.I.Manual of propaedeutic pediatrics Ukrmedkniga 2005 -218 p
2. Brennemann J. A contribution to our knowledge of the etiology and nature of hard curds in infants’ stools. Am. J. Dis. Child. 1911;1:341-359
3. Brennemann J. The care of infants. J. Am. Med. Assoc. 1912;59:542–543, 623–624, 720–721, 797–798
4. de Roos V. M., Katan M. B. Effects of probiotic bacteria on diarrhea, lipid metabolism, and carcinogenesis: a review of papers published between 1988 and 1998. Am. J. Clin. Nutr. 2000;71:405-411
5. Elwood P. C., Newton D., Eakins J. D., Brown D. A. Absorption of iron from bread. Am. J. Clin. Nutr. 1968;21:1162-1169
6. Epps R. P., Jolley M. P. Unsupervised early feeding of solids to infants. Med. Ann. D. C. 1963;32:493-495
7. Executive Board American Academy of Pediatrics: Committee on Nutrition, Scope and Functions. Pediatrics 1956;18:159
8. Fomon S. J. Nutrition of Normal Infants 1993 W. B. Saunders St. Louis, MO.
9. Fomon S. J., Filer L. J., Jr, Anderson T. A., Ziegler E. E. Recommendations for feeding normal infants. Pediatrics 1979;63:52-59


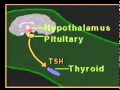{kind=link}