Заняття 1. nПредмет і завдання медичної генетики.
Предметом генетики людини є вивчення закономірностей nспадковості і мінливості у людини при всіх рівнях її організації та існування: nмолекулярному, клітинному, організменному, популяційному, біохронологіч-ному, nбіогеохімічному. Медична генетика вивчає роль спадковості в патології людини, nзакономірностей передачі від покоління до покоління спадкових захворювань, nрозробляє методи діагностики, лікування, профілактики спадкової патології, nвключаючи і захворювання зі спадковою схильністю.
 Синдром Елерса-Данлоса
Синдром Елерса-Данлоса
n
n Медична генетика вивчає:
n
n- значення спадкових факторів в етіології захворювань;
n
n- співвідношення спадкових і факторів середовища в генезі захворювань;
n
n- роль спадкових факторів у клінічній картині захворювання;
n
n- вплив спадковості на виздоровлення людини і наслідки захворювання;
n
n- вплив спадкових факторів на використання лікарських препаратів та інші види nлікування.
n
Історія медичної генетики.
В останні десятиліття значно nзмінилась структура захворювань населення. Захворювання з екзогенними факторами nетіології, як то інфекційні авітамінози, отруєння, відійшли на задній план nзавдяки значним досягненням мікробіології, імунології і біохімії, а на перший nплан виступили захворювання з ендогенними факторами етіології, тобто спадкові.
За даними експертів nВсесвітньої організації охорони здоров’я (ВООЗ), одна дитина із 100 nновонароджених страждає важким спадковим захворюванням внаслідок уродження nхромосом, у 4% дітей спостерігаються різні генетичні дефекти. Генетичні дефекти є також причиною 40% спонтанних nабортів. Кожна людина є носієм 15-20 потенційно дефектних генів.
Розроблення сучасних nбіохімічних, цитологічних і генетичних методів досліджень сприяло розкриттю nмолекулярної сутності багатьох захворювань. Було встановлено, що в розвитку як nспадкових, так і не спадкових (екзогенних) захворювань істотне значення має nстан генетичного апарату клітин організму. Сьогодні nгенетика є базовою для всіх біологічних наук, у тому числі й медичних.
Завданням сучасної медицини є поступовий перехід із сфери лікування nхворих у сферу запобігання хворобам і збереження здоров’я населення.
Значення основ медичної генетики потрібно не тільки лікарю, а й nсередньому медичному працівнику під час догляду за хворими і здійснення nзапобіжних заходів.
Уже в 1750 р. французький лікар П. Мопертюї описав характер nуспадкування багатополості (полідакталії). Проведений ним аналіз успадкування nцієї ознаки багато в чому передував відкриттю Г. Менделя. У 1814 р. Дж. Адамс nопублікував працю, в якій розрізняв не спадкові й спадкові захворювання.
За період 1803-1820 р. кілька лікарів описали тип успадкування nгемофілії. Швейцарський лікар-офтальмолог Й. Горнер у 1876 р. описав тип nуспадкування дальтонізму (колірної сліпоти). Російський лікар В. М. Флоринський nу монографії,, Вдосконалення і виродження людського роду” (1866 р.) писав, nщо збереження і покращення її можливе лише за доцільного підбору подружжя.
Генетика людини як наука виникла завдяки працям англійського вченого Ф. nГальтона (1822-1911 рр.). він разом із Г. Менделем є один із засновників nгенетики як науки. Гальтон вивчав успадкування розумових здібностей, nобдарованість, таланту людини, створив особливий напрям генетики – євгеніку, nпризначення якої – вдосконалити людину і людський рід.
Менделісти поч. ХХ ст. (У. Бетсон, В. Іогенсен) та ін. Вивчали якісні nознаки, які визначаються окремими генами і стверджували, що ці гени визначають nхарактер спадковості людини.
У 1918 р. Фішер довів, одні ознаки людини визначаються якістю, тобто nокремими генами, а другі – кількістю.
У 1900 р. К. Ланштейнер відкрив групи крові системи АВО і тим самим nзаклав початок вивченню поліморфних ознак людини.
У 1913 р. було описано поліморфізм відносно виявлення здібностей nвідчувати смак розчину фенілтіосечовини.
Лікар А. Гаррод (1902 р.) розробляв проблему порушення обміну речовин у nлюдини захворюванні алкаптанурією.
Алкаптанурія – спадкове захворювання, яке зумовлено неповноцінністю nферменту оксидази гемогентрозинової кислоти. А. Гаррод сформував знамените nположення про спадкові дефекту обміну речовин, тобто заклав основи біохімічної nгенетики.
Дж. Бідл і Є. Теймен, вивчаючи біосинтез тіаміну, встановили, що за nсинтез кожного ферменту відповідає певний ген. Вони виклали гіпотезу,, один ген n– один фермент”.
У 1908 р. Д. Харді, математик із кембріджського університету, і В. nВайнберг, лікар із Штутгарда, незалежно один від одного заклали основи nпопуляційної генетики і сформулювали закон, який носить їхнє ім’я. Закон nХарді-Вайберга було відкрито під час вивчення розподілу різних ознак у nпопуляцій людини.
У 20-х і 30-х рр. такі досліди, як Р. Фішер і Дж. Халдейн в Англії, С. nВайт у США, Г. Дольберг у Швеції, Л. Хогбен і Ф. Берштейн у Німеччині внесли nвеликий вклад теорію генетики і еволюції, в розробку статистичних методів вивчення nгенетики людини. Ці вчені описали методи аналізу закономірності успадкування, nрозщеплення, щеплення ознак і визначення частоти мутації.
Великий внесок у розробку проблем загальної генетики і генетики людини nв ті роки внесли вчені М. К. Кольцов, О. С. Себеровський, Ю. О. Філіпченко. nПрофесор С. Г. Левіт, який був керівником Московського Медико-генетичного nінституту до 1937 р. проводив цінні дослідження з генетики цукрового діабету.
Великий російський фізіолог І. П. Павлов дійшов до висновку, що треба nвивчати генетику для кращого знання фізіології.
У 1956 р. Д. Тійло і А. Леван встановили, що кількість хромосом у nсоматичних клітинах – 46, після чого були виявлені зміни хромосом при різних nзахворюваннях. І. Лежен у 1959 р. відкрив зайву 21-шу хромосому при хворобі nДауна.
У 1969 р. Т. Каперсон запропонував диференціальне фарбування хромосом, nщо дало змогу розрізняти кожну з хромосом окремо і виявляти зміни їх.
Великий внесок у вивчення загальної генетики людини зробили: М. П. nДубинін, Д. Д. Ромашов, А. А. Малиновський, В. П. Єфроїмсон, М. П. Бочков, І. nР. Барляк, М. А. Пілінський.
В Україні питаннями медичної генетики займалися такі відомі вчені, як nТ. І. Юдін, Б. М. Манківський.
Юдін займався питаннями євгеніки, а Манківський – лікуванням хворих nспадкову м’язову дистрофію і спадковими захворюваннями нервової системи.
Етапи nстановлення генетики
(1865 р.) n1900-1930 – період класичної генетики:
- створення теорії гена хромосомної теорії спадковості;
- формування уявлення про співвідношення генотипу та фенотипу, взаємодію генів, генетичні принципи індивідуального добору селекції;
- залучення генетичних ресурсів для цілей селекції.
n
1930-1953 nрр. – Період неокласичної генетики:
- робота в галузі штучного мутагенезу;
- виявлення того, що ген є складною системою, яка дробиться на частини.
- обґрунтування принципів популяційної генетики;
- створення біохімічної генетики, з’ясування ролі ДНК.
n
З 1953 р. n– епоха синтетичної генетики:
- розкриття структури та генетичної значущості ДНК;
- з’ясування на молекулярному рівні природи гена;
- початок робіт у галузі генної інженерії.
n
Спадковістю називається властивість повторювати в ряді поколінь nпотрібні ознаки і забезпечувати специфічний характер індивідуального розвитку в nпевних умовах середовища. Завдяки спадковості батьки і потомки мають подібний nтип біосинтезу, який визначає подібність у хімічному складі тканин, характері nобміну речовин, фізіологічних функцій, морфологічних ознак та інших nособливостей. Внаслідок цього кожний вид організмів відтворює себе із покоління nв покоління.
Мінливість – це явище, певною мірою протилежне спадковості, і виявляється в тому, nщо у будь-якому поколінні окремі особи чимось відрізняються і одна від одної, і nвід своїх батьків. Відбувається це тому, що властивості і ознаки кожного nорганізму – це результат взаємодії двох причин: спадкової інформації і nконкретних умов зовнішнього середовища, які можуть впливати як на зміну nспадкових задатків, так і на варіабельність виявлення їх.
Генетика як наука виникла внаслідок практичних потреб. При розведенні домашніх тварин і культурних рослин nздавна використовувалась гібридизація, порід, сортів і відрізняється один від nодного якими-небудь ознаками. Порівнюючи гібриди з вихідними формами, практики nдавно помітили деякі особливості успадкування ознак. Дарвін надавав великого nзначення вивченню закономірностей спадковості і мінливості і встановив, що вони nлежать в основі еволюції органічного світу.
На вивченні генетичних закономірностей ґрунтується селекція тобто nстворення нових і покращення існуючих порід домашніх тварин, сортів культурних nрослин, а також мікроорганізмів які використовуються у фармацевтичній nпромисловості, медицині і народному господарстві.
Велике значення має генетика для медицини ветеринарії оскільки багато хвороб людини і тварини спадкові і nдля лікування їх або запобігання потрібні генетичні досліди.
Основні закономірності успадкування властивостей і nознак були відкриті Г. Менделем (1822-1884). Однак ці дослідження не були зразу належно оцінені і nзалишалися мало відомими до 1900 р., коли водночас три дослідники (Г. де Фріз у nГолландії, Т. Корренс у Німеччині, Є. Чермак в Австрії) незалежно один від nодного вдруге відкрили закони спадковості, сформульовані Менделем. Цю дату nвважають датою створення експериментальної генетики.
При вивченні закономірностей успадкування звичайно схрещують організми, nщо відрізняються один від одного альтернативними, тобто контрастуючими проявами nознаки. Наприклад можна взяти горох (саме його брав Мендель) з насінням жовтого nі зеленого кольорів (ознака – колір насіння), зморшкуватим і гладеньким (ознака n– форма насіння), забарвленням квіток пурпуровим і білим (ознака – колір nквітки), з високим і низьким стеблом (ознака – розмір стебла).
Кожна ознака організму визначається одним або кількома генами. Кожний nген може існувати в кількох формах (станах), які називають алелями n(алеломофорними парами). Алелі гена розташовані у гомологічних хромосомах в nодних і тих самих місцях (локусах).
Гомозиготи і гетерозиготи
Якщо в обох nгемологічних хромосомах містяться однакові алелі (наприклад, обидва кодують nжовте забарвлення насіння або обидва – зморшкувату форму насіння тощо), такий nорганізм називається гомозиготним. Якщо ж алелі різні (наприклад в одній із nгомологічних хромосомах алель кодує жовтий пігмент, а в іншій, гомологічній їй, nхромосомі алель зеленого пігменту або один алель – гладенької форми, а другий – nзморшкуватої форми насіння), то такий організм називається гетерозиготним.
Можна сказати і так: зигота, яка утворилась злиттям гамет з однаковими nалелями одного гена, називається гемозиготною. Гетерозигота утворюється злиттям nгамет, які несуть у собі різні алелі даного гена. Один і той самий організм nможе бути гомозиготним за одним (або кількома) генами (вв, ББ) і гетерозиготним nза іншим (іншими: Аа, Гг).
Генотип і фенотип
Сукупність спадкових факторів організмів (генів) називається генотип. nСукупність всіх ознак і властивостей організму, які є результатом взаємодії nгенотипу з зовнішнім середовищем називається фенотипом. Ось чому організми з nоднаковим генотипом можуть відрізнятися один від одного залежно від умов nрозвитку і існуванням. Межі, в яких змінюються фенотипові прояви генотипу, nназиваються нормою реакції.
Актуальні проблеми генетики
Генетика – наука, що об’єднує навколо своєї nпроблематики багато біологічних дисциплін.
Біохімічна генетика включає біохімію nнуклеїнових кислот, білків і ферментів. Тут застосовуються методи, що nвикористовуються біохіміками і молекулярними біологами (хроматографія, аналіз nферментів).
Цитогенетика займається вивченням хромосом nтварин і рослин в нормі і при патології.
Класична генетика розглядає успадкування Менделевських ознак і з nдопомогою статистичних методів досліджує більш складні типи успадкування.
Клінічна генетика вирішує питання діагностики, прогнозування і nлікування різних спадкових хвороб.
Предмет і nзавдання генетики
Популяційна генетика вивчає поведінку генів в популяціях і дію таких nфакторів, як дрейф генів, міграції, мутації і добір.
Генетика nповедінки – наука, предметом вивчення якої являються спадкові фактори, що nвизначають поведінку людей.
Соціальна біологія пояснює поведінку людини в суспільстві на сонові nбіологічних і еволюційних уявлень.
Завдання nгенетики
1. Одне з головних завдань сучасної біології – nзбільшення ресурсів для населення Землі, що постійно зростає.
Генетика являється nтеоретичною основою селекції, що розробляє ефективні шляхи і методи одержання nнових порід тварин і сортів рослин. Найважливішим інструментом селекції став nзакон гомологічних рядів, відкритий М. І. Вавиловим.
Генетики-селекціонери nвикористовують такий природний процес, як мутагенез, примушуючи його служити nлюдині. Головна задача селекції майбутнього – одержати спрямовані мутації тобто nнавчитися стріляти по потрібному гену. При цьому використовують радіоактивне nвипромінювання і хімічні мутагени.
2. Генетика людини більшістю своїх досягнень зобов’язана nтому, що опиралась на закони Менделя, і використовувала методи, що розроблялися nв різних областях біології (регуляція активності генів, регуляція діяльності nімунної системи і роботи мозку, причини спадкових хвороб і т. д.).
3. Пізнання молекулярних основ життєдіяльності nорганізмів призвело до використання біологічних процесів і речовин в промисловості. nНародилася нова галузь виробництва – біотехнологія, що являє собою комплект nбіологічних знань і технічних засобів, які необхідні для одержання продуктів nжиттєдіяльності клітини.
Основні напрямки nсучасної біотехнології – біотехнологічний синтез, культивація і використання nрослин і клітинних тварин, генна інженерія, наука про білкові речовини клітин n(ензімологія).
Як nрізноманітна спадковість людини за своєю організацією і функціями, так і nчисленні її патологічні варіації. В основі різноманітності реакцій nлюдини на зовнішні фактори лежить генетичний поліморфізм, коли одна і та ж nознака може детермінуватись різними генами, різними алелями одного гена. Коли nзахворювання викликається багатьма факторами зовнішнього середовища, nполіморфізм виражений більше. Захворювання зі спадковою схильністю визначається nпоєднанням спадкових і зовнішніх факторів, що дозволяє віднести їх до nзахворювань з пенетрантністю, яка в значній мірі залежить від умов середовища.
n nЗначна питома вага природженої та спадкової патології в захворюваності і nсмертності людини, надзвичайно негативний вплив даної патології як на окрему nлюдину, так і на популяцію в цілому, приводить до погіршення демографічних nпоказників.
n Розвиток медико-генетичної nслужби є першочерговим завданням сучасної медицини. На даний nчас в області медико-генетичного консультування діє наказ № 77 від 14.01.93. n“Про стан та заходи подальшого розвитку медико-генетичної допомоги в Україні”. nГотуються зміни та удосконалення даного наказу. Згідно наказу в Україні діють n21 обласні медико-генетичні консультації (МГК), по 3-4 в кожній області, nміжрайонні МГК, а також 7 ММГЦ (міжобласних медико-генетичних центрів) (Київ, nЛьвів, Донецьк, Кривий Ріг, Одеса, Харків), включая 1 Республиканську nММГЦ–республікиКрим.
n nНа даний час Україна відноситься до країн з від’ємним природним приростом nнаселення. Спадкові захворювання і природжені вади розвитку (ПВР) в структурі nзахворюваності дітей першого року життя становлять 2,9%, смертності 31%, у nструктурі поширеності захворювань серед дітей 0-14 років 1,27%, у структурі nінвалідності 19,7%, смертності 20%. Захворюваність новонароджених на природжені nвади розвитку і спадкові захворювання з 1990 року збільшилось в 1,3 рази, у nдітей від 0 до 1 року в 2,1 рази, від 0 до 14 років в 1,8 рази.
n У нас в nобласті за період з 1988 по 1998 рр. частота ПВР складає 3,2%, в 1996-1998 – nзбільшилось до 3,5%, в останні роки ~ 2,6-2,7%.
n nАле частота основних ПВР, що сприяють інвалідності і смертності nутримується на попередніх цифрах з тенденцією до зростання за рахунок nпокращення діагностики даної групи патології (вади серцевої системи) тощо.
n Частота nПВР, що стали причиною смерті у дітей першого року життя складає 46,6 за період n1988-1998 р.р. в останні роки коливається від 39 до 44 на 10 000 населення, nзаймає, як правило, друге місце, а в окремі роки (1990, 1996, 2000 рр.) і перше nмісце серед інших причин. За 2002 рік показник знизився до 10,4%, частота його n28,8 на 10 000 населення.
n
n Основні завдання медико-генетичних установ та принципи nмедико-генетичного консультування:
n
n– Верифікація або встановлення діагнозу ПВР або спадкових захворювань, nдуже часто синдромального діагнозу.
n
n- Визначення ризику виникнення даної патології в конкретній сім’ї в математичному nвиразі, допомога в прийнятті правильного рішення щодо подальшого народжування дітей.
n
n- Проведення заходів, спрямованих на попередження народжування хворих дітей – nретроспективного (в сім’ях, де є хворі) і – проспективного характеру n(профілактика, де немає хворих).
n
n Основні контингенти хворих, що підлягають консультації nв обласній МГК.
n
n1. Діти з множинними вродженими вадами розвитку для виключення можливості nсиндромальної патології.
n
n2. Діти з ізольованими вадами розвитку.
3. nДіти з розумовою відсталістю неуточненої етіології при спокійному nперинатальному анамнезі та прогредієнтному перебігу захворювання для виключення nспадково детермінованої патології обміну речовин.
n
n4. Діти з затримкою у фізичному розвитку в поєднанні з диспластичним фенотипом, nмножинні стигми дизембріогенезу.
n
n5. Хворі з аномаліями статевої диференціації.
n
n6. Хворі з передчасним та запізнілим статевим розвитком.
n
n7. Діти з ранньою маніфестацією бронхолегеневої патології в поєднанні nкишково-шлунковими розладами, тяжкою гіпотрофією для виключення муковісцидозу nта інших симптомів мальабсорбції.
n
n8. Подружні пари з тривалим непліддям (2 роки і більші років).
n
n9. Подружні пари, в яких є або були діти з ПВР та спадковою патологією n(моногенною, хромосомною, мультифакторною) для визначення прогнозу майбутніх nнащадків.
n
n10. Хворі первинною та вторинною аменореєю.
n
n Усі nгенотипові ознаки людини, як і інших живих істот є генетично обумовленими. nВідповідно спадкова інформація про них закладена в генах. Сукупність усіх nгенів, притаманних організму, називається генотипом. Внаслідок реалізації nгенетичної інформації, закладеній в генотипі, формується фенотип – сукупність nзовнішніх ознак та властивостей організму.
n Відомо, nщо основним носієм спадкових ознак є ДНК, яка знаходиться в ядрі клітини
n nВ інтерфазному ядрі (яке не ділиться) весь спадковий матеріал nпредставлений нитками хроматину. Підчас поділу (мітозу) він представлений nхромосомами (23 парами). 46, ХХ – жінка. 46, ХY – чоловік.
n
n nДНК – це послідовність нуклеотидів. Триплет нуклеотидів – код певної амінокислоти. Певна послідовність nамінокислот – це той чи інший білок, а кодує його відповіддю ген. Таким nчином, певна послідовність нуклеотидів, яка кодує певний білок (фермент), якусь nознаку, є ген. Сучасне визначення гену n(Лондон, 1892 рік): ген – це транскриптон – фрагмент молекули ДНК, який nвважається єдиним блоком, з прилягяючими до нього сервісними і контролюючими nпослідовностями нуклеотидів і кодує інформацію про послідовність амінокислот в nмолекулі поліпептидного ланцюга, оскільки містить інтрони.
n Нуклеотид – це азотиста основа – nаденін, тімін, гуанін, цитозін (в РНК замість тіміну – урацил) + залишок nфософорної кислоти і цукор, дезоксирибоза (в ДНК) і рибоза (в РНК). Отже: nосновними носіями ДНК, генів і відповідно спадкової інформації є хромосоми, які nрозміщуються в ядрі кожної клітини і видимі за допомогою оптичного мікроскопу. nКількість хромосом у кожного організму є специфічною, у людини вона складає 46 nу всіх клітинах, за винятком статевих, в тому числі 44 – аутосоми, дві – nстатеві хромосоми, які в цілому формують диплоїдний хромосомний набір, nпритаманний соматичним клітинам людини.
n nУ жіночому каріотипі міститься дві однакові статеві Х-хромосоми, а в nчоловічому – статеві Х- та Y-хромосоми. Власне Y-хромосома визначає розвиток в nчоловічому напрямку. За винятком статевих хромосом у чоловіків, решту 44 nаутосоми і статеві хромосоми жінок представлено парами хромосом з однаковим nнабором генів (гомологічні хромосоми). Кожна гомологічна хромосома походить від nбатька і матері. Чоловічі статеві хромосоми відрізняються набором генів і тому nє не гомологічними: Y-хромосому завжди успадковують від батька, а Х-хромосому nвід матері. У статевих клітинах є половинний (гаплоїдний) набір хромосом, який nскладається з 22 аутосом (по одній з кожної гомологічної пари) та однієї nстатевої хромосоми Х або Y.
n nХромосома – це макромолекула ДНК з генами, розміщеними в лінійному nпорядку, яка разом із гістоновими та негістоновими білками формує специфічну nспадкову структуру, здатну до самовідтворення. В інтерфазі, коли відбувається nфункціонування (експресія) генів, хромосома набуває вигляду розтягнутої нитки, nзавдяки чому забезпечується доступ регуляторів експресії до будь-якого гена. nПід час реплікації відбувається подвоєння геному за рахунок утворення другої nідентичної макромолекули ДНК. Це стосується всіх хромосом клітини. Хромосоми nпісля реплікації набувають двониткової (двохроматидної) структури. Під час nмітозу відбувається розходження хроматид у дочірні клітини, які утворюються з nматеринської. Таким чином, досягається розподіл генетичної інформації та її nсамовідтворення у поколіннях клітин.
n
n
Хромосоми, які перебувають у nінтерфазі, є недосяжними для спостереження у світловому мікроскопі. У мітозі nвони скорочуються в довжину і зі стадії профази стають видимими у світловому nмікроскопі. Найбільш контурованого стану хромосоми набувають під час середньої nметафази. Тому в науці та практичній медицині аналіз кількості та структури nхромосом під світловим мікроскопом побудований здебільшого на аналізі в період nсередньої метафази. Генотип – це не просто сума nнезалежних генів, а система генів, яка базується на їх постійній взаємодії. nПроцес взаємодії генів відбувається внаслідок взаємодії кодованих ними nпродуктів – поліпептидів і білків. Ген контролює розвиток ознаки через низку nпроміжних ланок, які в свою чергу контролюються генетично. Найважливішими nпроміжними етапами функціонування кожного гена є транскрипція і трансляція n(синтез білка), які здійснюються ферментами, рибосомальними РНК, транспортними nРНК, амінокислотами, які запрограмовані генетично. Структурні, транспортні nбілки і ферменти, взаємодіючи, утворюють складніші структури організму – від nклітини до органа. Узгоджена робота різних генів лежить в основі програми nформування фенотипу і, таким чином, є основою для нормальних чи патологічних nпроцесів в організмі.
nВажливим чинником взаємодії генів є гормони. Щоб вплинути на клітину-мішень, nгормон вступає в комплекс з рецепторним білком, утворення якого також генетично nдетерміноване. Утворений комплекс транспортується в ядро клітини, де активізує n(експресує) або пригнічує (репресує) функціонування генів. Таким чином, завдяки nвзаємодії генів на різних рівнях досягається точна і швидка регуляція реакції nорганізму, яке відбувається внаслідок зміни внутрішнього середовища і зовнішніх nчинників.
nОднак визначальним для формування ознак і властивостей організму є взаємодія nпар алельних генів.
n Кожна nлюдина отримує половину хромосом від батька, а половину від матері, тому nгенетичний матеріал є продубльованим. У геномі кожний ген представлений двома nалелями, які займають відповідні одна одній ділянки на гомологічних хромосомах.
n nАлель – це чітко визначений варіантний стан гена, який може бути nнормальним або мутантним. Алелі можуть розрізнятися між собою за первинною nпослідовністю нуклеотидів, але всі алелі (варіанти) одного гену формують nреалізацію тієї самої ознаки.
nЯкщо материнська і батьківська гомологічні хромосоми несуть однакові алелі – це nгомозиготне носійство генів, якщо алелі різні, це гетерозиготне носійство гена. nЗ двох алелей (материнського і батьківського) одного гена у разі nгетерозиготного носійства проявляється ефект лише одного алеля. Це явище nназивається домінуванням, а ознака, для прояву якої достатньо лише одного алеля n– домінантною. Присутність ознаки за умови наявності двох однакових алелей nсвідчить про явище рецесивності, таку ознаку називають рецесивною. Рецесивною nознакою є, наприклад, забарвлення райдужної оболонки ока у блакитний колір, а nдомінантною – в карий.
nЯвище домінантності –рецесивності проявляється у всіх випадках, коли ознака nвизначається генами, розташованими у гомологічних хромосомах. Воно властиве nгенам, розміщених у аутосомах і Х-хромосомах жіночого каріотипу.
n Що стосується статевих хромосом nчоловіків , то Х- і Y- хромосоми не є гомологічним і містять у своєму складі nрізний набір генів. Тому у чоловіків проявляються всі ознаки, кодовані nХ-хромосомою, навіть ті, які у жінок можуть проявлятись лише у гомозиготному nстані, тобто рецесивні. Це положення є визначальним для розуміння nетіопатогенезу Х-зчепленої патології людини.
n
nЗгадане явище домінантності–рецесивності проявляється в основному на nвізуальному (клінічному рівні). Що стосується біохімічних параметрів, особливо nна рівні синтезу білку, то найчастішим є кодомінантний прояв ознаки. У такому nразі проявляється функціональна активність обох алелей у процесі синтезу білка. nПересвідчитись в цьому можна, здійснівши електрофоретичне обстеження: якщо nалелі різні, то наявні дві окремі фракції білка, які відрізняються за nелектрофоретичною рухливістю. Кодомінантний прояв властивий великій кількості nнормальних ознак людини. Класичним прикладом є система груп крові, зокрема АВ0, nколи еритроцити несуть антигени, контрольовані обома алелями одного гена.
nВсі спадкові хвороби можна поділити таким чином:
n nІ. Хромосомні – внаслідок зміни числа (цифрові аберації) або структури n(стуктурніаберації).
n
nПриклади:47,ХХ(+21)–хворобаДауна.
n
n47,ХY(+18)–синдромЕдварса.
n
n47,ХХ(+13)–синдромПатау.
n45,ХО–синдромШерешевського-Тернера
47,ХХY–синдромКлайнфельтера.
n
n46,ХХ(5р-)–синдром“котячогокрику”
n
n46,ХY(4р-)синдромВольфа-Хіршхорна.
n
n ІІ. nМоногенні хвороби – це хвороби, коли зміна коду на рівні порушення nпослідовності нуклеотидів – точкова мутація, зумовлює цілий комплекс відхилень nв організмі. Ланки патогенезу генних захворювань: мутантний алель – nпатологічний продукт (якісно або кількісно), ланцюг наступних біохімічних nпроцесів – клітини – органи – організм. Особливості клініки генних захворювань: nклінічний поліморфізм, вікова варіабельність маніфестації, прогредієнтність nклінічної картини та хронічний перебіг, явища антиципації (наростання важкості nперебігу у наступних поколіннях).
Найбільш обширна і вивчена група nмоногенних захворювань – це ензимопатії. Як відомо, всі біохімічні процеси nу живих організмів знаходяться під генетичним контролем. Окремий біохімічний nпроцес складається із цілої серії послідовних реакцій, кожна з яких контролюється nокремим ферментом. Будь-який з генів, відповідальних за утворення ферментів, nможе піддаватися мутації, в результаті якої фермент або змінює свою структуру і nфункціональні властивості, або зовсім не утворюється, і тоді реакція, що nпротікає при його участі, блокується. Блок може виникнути в будь-який із ланок nланцюга біохімічної реакції. В результаті такого блоку може мати місце:
n
n- недостатнє утворення продуктів даної реакції і більш віддалених продуктів nйого перетворення;
n
n- нагромадження в організмі субстрату блокованої реакції або його попередників;
n
n- зміни основного напряму в перебігу реакції і підвищене утворення продуктів, nякі в нормі є в незначних кількостях.
n
n Класичний nпрояв біохімічних мутацій у людини не завжди пов’язаний з первинним ефектом дії nгенів. Як правило, розвиваються супутні вторинні порушення хімізму і функції nтканин, які інколи по яскравості патологічних проявів виступають навіть на nпередній план. Це явище відоме в генетиці, як множинний ефект або плейтропізм nгенів. Вторинні зміни не базуються безпосередньо на дефекті гена і не пов’язані nіз структурними змінами білка. Вони винятково є результатом зсуву обміну nречовини, який настає під впливом первинного порушення, і може nрозповсюджуватись на декілька біохімічних реакцій в різних тканинах, набуваючи nрізноманітних проявів в залежності від специфіки цих тканин. Так, при nфенілкетонурії відсутність ферменту, який розщеплює фенілаланін до тирозину, nприводить до нагромадження фенілаланіну в тканинах і перетворення його в nтоксичні для нервової системи кислоти. Це веде до розумової відсталості дитини. nНедостатня кількість проміжних продуктів нормального обміну тирозину призводить nдо зменшення утворення адреналіну і меланіну і виявляється у дітей в зменшенні nпігментації, артеріальної гіпотонії. Діагностичне значення плейотропії полягає nв тому, що клінічно встановлений симптом є важливим показником інших, перш за nвсе прихованих патологічних проявів.
n 1. nАутосомно-домінантний тип успадкування: передача від батьків до дітей по nвертикалі, пенетрантність – фенотипичні прояви у носія патологічного гену, і nекспресивність патологічного гену (ступінь вираженості ознак).
nПриклади:
n
n-нейрофіброматозРекленгаузена,
n
n-синдромМорфана,
n
n-синдромАперта,
n
n-синдромФранческлетті.
n2. Аутосомно-рецесивний тип успадкування: батьки фенотипово здорові.
n Приклади МВ, більшість захворювань обміну nречовин (ензимопатії), фенілкетонурія, лейкодистрофії, нейроліпідози, тощо, nсиндром МПВР – Секкеля,Меккеля.
n
n3.Х-зчеплені–домінантні-фосфат-діабет,
n
n-рецесивні–гемофілія,дальтонізм,Х-зчепленийімунодефіцит.
n
n4. Моногенні захворювання з неуточненим типом успадкування: синдром nРосела-Сільвестра, синдром обличчя Ельфа.
nІІІ. nМультифакторіальні захворювання. До таких хвороб відносяться всі гіпертензії n(6-8 генів), сімейну гіперхолестеринемію (6 генів), порушення імунітету (32 nгени), цукровий діабет (8 генів, пара з яких рецисивні), тромбози (6 генів), nбронхолегеневі хвороби (6 генів), шизофренія, епілепсія, депресивні синдроми n(6-8 генів), дефекти обміну речовин (від 2 до 10 генів), ішемічна хвороба nсерця, ожиріння.
n Подібно до того, як спадковість nлюдини різноманітна за своєю організацією і функціями, численні її патологічні nваріації. В основі різноманітності реакції людини на зовнішні фактори лежить nгенетичний поліморфізм, коли одна і таж ознака може детермінуватись різними nгенами, різними алелями одного гена. Коли хвороба викликається багатьма nфакторами зовнішнього середовища, поліморфізм виражений ще більше. Хвороби із nспадковою схильністю і визначаються поєднанням спадкових і зовнішніх факторів, nщо дозволяє віднести їх до захворювань з пенетрантністю, яка в значній мірі nзалежна від усіх умов середовища. Поєднане вивчення каріотипу людини, фізичних nі фізіологічних особливостей організму, біохімічні дослідження в сукупності з nпопуляційним і генеалогічним аналізом відкривають широкі перспективи для nклінічної і профілактичної медицини.
n МПВР – це nє комплекс вад розвитку, коли вади стосуються двох органів, що належать до nрізних систем, їх не більше двох, вони не індукуються одна одною.
n nСеред них також відмічають хромосомні, моногенні, тератогенні комплекси nі МПВР з неуточненою етіологією (некласифіковані комплекси). Ця група nнайскладніша для діагностики і консультування. Сучасний каталог В. Мак К’юсика nналічує біля 10 000 синдромів. Велике значення має правильне описання фенотипу n(тому необхідно направляти на консультацію жінку, якщо вона мала nМПВР плоду, ретельно описати фенотип, бажано подробний протокол nпатологоанатомічного розтину. Правильний синдромальний діагноз дає можливість nвстановити тип успадкування, запропонувати заходи по його попередженню. В nсучасних умовах розповсюджені діагностичні комп’ютерні програми, але вони також nвимагають знання генетики, тому що виділяють декілька синдромів, тобто звужують nколо пошуку, але все одно потрібний аналіз лікаря.
n
n
nСпадкові аномалії – це велика група вад розвитку, nпри яких дефект закладений в генах і передається по спадковості.
nII. Вроджені аномалії – це вади nрозвитку, які плід набуває під час внутрішньоутробного розвитку.
nСпадкові ( nхромосомні, генні).
n Зумовлені впливом шкідливих факторів зовнішнього середовища (травми, опіки).
Зумовлені nспадковою схильністю і дією провокуючих факторів зовнішнього середовища n(цукровий діабет, виразкова хвороба, пухлини).
Хромосомні-обумовлені порушеннями кількості і структури хромосом.
Генні-обумовлені порушенням в структурі генів.
Хромосомні хвороби.
nАномалії nстатевих хромосом.
– Синдром Клайнфельтера
– nсиндром nШерешевського-Тернера
– nсиндром nтрисомії Х
Аномалії аутосом:
Синдром nДауна
Синдром Едвардса
Синдром Патау
Хвороби амінокислотного обміну (фенілкетонурія, nгомоцистінурія, гістидінемія).
Спадкові дефекти обміну вуглеводів (галактоземія, nфруктоземія, глікогенози, гіпо- і алактазія).
Порушення ліпідного обміну (ліпідози).
nПорушення nобміну мукополісахаридів (мукополісахаридози).
Генеалогічний nметод вивчення спадковості людини
Основний nметод генетичного аналізу в людини полягає у складанні і вивченні родоводу.
Генеалогія n- це родовід. Генеалогічний метод – метод родоводів, коли простежується ознака n(хвороба) в родині з указанням родинних зв’язків між членами родоводу. В його nоснову покладено ретельне обстеження членів родини, складання й аналіз родоводів.
Це nнайбільш універсальний метод вивчення спадковості людини. Він і nвикористовується завжди лри підозрі на спадкову патологію, дозволяє встановити nу більшості пацієнтів:
• nспадковий характер ознаки;
• тип nуспадкування і пенетрантність алеля;
• nхарактер зчеплення генів і здійснювати картування хромосом;
• nінтенсивність мутаційного процесу;
• nрозшифрування механізмів взаємодії генів.
Цей метод nзастосовують при медико-генетичному консультуванні.
Суть генеалогічного nметоду полягає у встановленні родинних зв’язків, простеженні ознак або хвороби nсеред близьких і далеких, прямих і непрямих родичів.
Він nскладається із двох етапів: складання родоводу і генеалогічного аналізу. nВивчення успадкування ознаки або захворювання в певній сім’ї розпочинається з nсуб’єкта, який має цю ознаку або захворювання.
Особина, nяка першою попадає в поле зору генетика, називається пробандом. Це переважно nхворий або носій дослідної ознаки. Діти однієї батьківської пари називаються сибсами nпробанда (брати – сестри). Потім переходять до його батьків, далі до братів і nсестер батьків і їх дітей, потім до дідусів і бабусь і т.д. Складаючи родовід, nроблять короткі нотатки про кожного з членів сім’ї, його родинні зв’язки з nпробандом. Схема родоводу (рис. 1) супроводжується позначеннями під рисунком і nотримала назву легенди.
Рис. 1. Родовід сім’ї, nде успадковується катаракта:
хворі на цю недугу – nчлени родини I – 1, IІ – 4, III – 4,
Застосування генеалогічного методу дозволило встановити характер nуспадкування гемофілії, брахідактилії, ахондроплазії та ін. Він широко nвикористовується для уточнення генетичної природи патологічного стану і при nскладанні прогнозу здоров’я нащадків.
Методика nскладання родоводів, аналіз. Складання родоводу розпочинають з пробанда – nлюдини, яка звернулася до генетика або до лікаря і містить ознаку, яку nнеобхідно вивчити у родичів по батьківській і материнській лініях.
При nскладанні родовідних таблиць користуються умовними позначеннями, nзапропонованими Г. Юстом у 1931 р. (рис.2). Фігури родоводів розміщують nгоризонтально (або по колу), в один рядок кожне покоління. Зліва позначають nримською цифрою кожне покоління, а окремих осіб у поколінні – арабськими зліва nнаправо і зверху вниз. Причому найстарше покоління розташовують зверху родоводу nі позначають цифрою І, а наймолодше – внизу родоводу.
Рис. 2. Умовні nпозначення, які використовуються при складанні родоводів.
Братів і nсестер згідно з народженням найстаршого розташовують зліва. Кожний член nродоводу має свій шифр, наприклад, II – 4, IIІ – 7. Шлюбна пара родоводу позначається за тим же nномером, але з малої літери. Якщо один із подружжя необстежений, відомості про nнього не наводяться взагалі. Всі індивідууми розміщуються строго по поколіннях. nЯкщо родовід великии, то різні покоління розташовуються не горизонтальними nрядами, а концентричними.
Після nскладання родоводу до нього додається письмове пояснення – легенда родоводу. У nлегенді мають знайти віддзеркалення такі відомості:
• nрезультати клінічного і позаклінічного обстеження пробанда;
• відомості nпро особистий огляд родичів пробанда;
• nзіставлення результатів особистого огляду пробанда з відомостями опитування nйого родичів;
• nписьмові відомості про родичів, які проживають в іншій місцевості;
• nвисновок щодо типу успадкування хвороби або ознаки.
Не слід nобмежуватися при складанні родоводу тільки опитуванням родичів – цього nнедостатньо. Частині з них призначають повне клінічне, позаклінічне або nспеціальне генетичне обстеження.
Мета nгенеалогічного аналізу полягає у встановленні генетичних закономірностей. На nвідміну від інших методів, генеалогічне обстеження повинно завершуватися nгенетичним аналізом його результатів. Аналіз родоводу дає можливість дійти nвисновку щодо характеру ознаки (спадкова чи ні), титл, успадкування n(аутосомно-домінантний, аутосомно- рецесивний або зчеплений зі статтю), nзиготності пробанда (гомо- або гетерозиготний), ступеня пенетрантності й nекспресивності досліджуваного гена
Особливості nродоводів при різних типах успадкування: аутосомно-домінантних, nаутосомно-рецесивних і зчеплених зі статтю. Аналіз родоводів показує, що всі nхвороби, детерміновані мутантним геном, підпорядковуються класичним законам nМенделя за різних типів успадкування.
За nаутосомно-домінантного типу успадкуванню домінантні гени фенотипно виявляються nв гетерозиготному стані і тому визначення їх, і характер успадкування не nвикликає утруднень.
Цьому nтипу успадкування властиві такі закономірності:
1) у nкожного ураженого хворий один із батьків;
2) в nураженого, який перебуває у шлюбі зі здоровою жінкою, в середньому половина nдітей хворіє, а друга половина – здорова;
3) у nздорових дітей ураженого одного з батьків діти й онуки здорові;
4) nчоловіки та жінки уражуються однаково часто;
5) nзахворювання повинно проявлятися в кожному поколінні;
6) nгетерозиготні індивідууми уражені.
Прикладом nаутосомно-домінантного типу успадкування може бути характер успадкування nшестипалості (багатопалості). Шестипалі кінцівки – явище досить рідкісне, але nстійко зберігається у багатьох поколіннях деяких родин (рис. 3). Багатопалість стійко повторюється в нащадків, якщо nхоча б один із батьків багатопалий, і відсутня в тих випадках, коли в обох nбатьків кінцівки нормальні. У нащадків багатопалих батьків ця ознака присутня в nрівній кількості у хлопчиків і дівчаток. Дія цього гена в онтогенезі nз’являється досить рано і має високу пенетрантність.
Рис. 3. Родовід при аутосомно-домінантному типі nуспадкування.
При аутосомно-домінантному типі успадкування ризик появи nхвороби в нащадків, незалежно від статі, складає 50 %, але прояви захворювання nпевною мірою залежать від пенетрантності.
Аналіз nродоводів показує, що за таким типом успадковуються: синдактилія, хвороба nМарфана, ахондроплазія, брахідактилія, геморагічна телеангіектазія Ослера, nгемахроматоз, гіпербілірубінемія, гіперліпопротеїнемія, різні дизостози, nмармурова хвороба, незавершений остеогенез, нейрофіброматоз Реклінгаузена, nотосклероз, хвороба Пельціуса – Мерцбахера, пельгірівська аномалія лейкоцитів, nперіодична адинамія, перніциозна анемія, полідактилія, порфирія гостра nінтермітуюча, птоз спадковий, ідіопатична тромбоцитопенічна пурпура, таласемія, nтуберозний склероз, фавізм, хвороба Шарко-Марі, хвороба Штурге-Вебера, множинні nекзостози, ектопія кришталика, еліптоцитоз (Л. О. Бадалян зі співавт, 1971).
За nаутосомно-рецесивного успадкування рецесивні гени фенотипно виявляються тільки nв гомозиготному стані, що затруднює як виявлення, так і вивчення характеру nуспадкування.
Цьому типу nуспадкування властиві такі закономірності:
1) якщо nхвора дитина народилася у фенотипно нормальних батьків, то батьки обов’язково nгетерозиготи;
2) якщо nуражені сибси народилися від близько-родинного шлюбу, то це доказ рецесивного nуспадкування захворювання;
3) якщо nвступають у шлюб хворий на рецесивне захворювання і генотипно нормальна людина, nвсі їх діти будуть гетерозиготами і фенотипно здорові;
4) якщо nвступають у шлюб хворий і гетерозигота, то половина їх дітей будуть уражені, а nполовина — гетерозиготні;
5) якщо nвступають у шлюб двоє хворих на одне те ж рецесивне захворювання, то всі їх nдіти будуть хворі.
6) nчоловіки і жінки хворіють з однаковою частотою:
7) nгетерозиготи фенотипно нормальні, але є носіями однієї копії мутантного гена;
8) уражені nіндивіди гомозиготні, а їх батьки – гетерозиготні носії.
Аналіз nродоводів свідчить, що фенотипне виявлення рецесивних генів відбувається тільки nв тих сім’ях, де ці гени мають обоє батьків хоча би в гетерозиготному стані n(рис.4). Рецесивні гени в людських популяціях залишаються nневиявленими.
Рис. 4. Родовід при аутосомно-рецесивному типі успадкування.
Проте у nшлюбах між близькими родичами або в ізолятах (невеликі групи людей), де nвідбуваються шлюби за близьких родинних зв’язків, прояв рецесивних генів nзростає. За таких умов ймовірність переходу в гомозиготний стан і фенотинного nвиявления малопоширених рецесивних генів різко зростає.
Оскільки nбільшість рецесивних генів має негативне біологічне значення й зумовлює nзниження життєвої стійкості та появу різних виродливосте і спадкових хвороб, то nдля здоров’я нащадків родинні шлюби мають різко негативний характер.
Спадкові nхвороби переважно передаються за аутосомно-рецесивним типом, діги від nбатьків-гетерозигот можуть успадкувати хворобу в 25 % випадків (при повній nпенетрантності). Зважаючи, що повна пенетрантність зустрічається рідко, то й nвідсоток успадкування захворювання менший.
За nаутосомно-рецесивним типом успадковуються: агаммаглобуліпемія, агранулоцитоз, nалкаптонурія, альбінізм (рис.5), амавротична nідіотія, аміноацидурії, анемія аутоімунна гемолітична, анемія гіпохромна nмікроцитарна, аненцефалія, галактоземія, гермафродитизм (рис.6), гепагоцеребральна дистрофія, хвороба Гоше, nєвнухоїдизм, мікседема, серпоподібноклітинна анемія, фруктозурія, кольорова nсліпота (Л. О. Бадалян із співавт., 1971).
Рис. 5. – Успадкування за аутосомно-рецесивним типом. nАльбінізм.
Рис.6. Успадкування за аутосомно-рецесивним типом. nГермафродитизм.
Низка nзахворювань успадковується за Х-хромосомним (зчепленим зі статтю) типом, коли nмати є носієм мутантного гена, а половина її синів хворі. Розрізняють nХ-зчеплене домінантне і Х-зчеплене рецесивне успадкування.
Родовід nХ-зчепленого домінантного успадкування (рис. 7). Для цього типу успадкування характерно:
1) nуражені чоловіки передають своє захворювання дочкам, але не синам;
2) nуражені гетерозиготні жінки передають захворювання половині своїх дітей nнезалежно від їх статі;
3) nуражені гомозиготні жінки передають захворювання всім своїм дітям.
Такий тип nуспадкування зустрічається не часто. Захворювання у жінок перебігає не так nтяжко, як у чоловіків. Досить важко розрізнити між собою Х-зчеплене домінантне nй аутосомно-домінантне успадкування. Застосування нових технологій (ДНК-зонди) nдопомагає більш точно виявити тип успадкування.
Рис. 7. Х-зчеплене домінантне успадкування.
Родовід nХ-зчепленого рецесивного успадкування (рис.8). Цьому типу успадкування властиві такі закономірності:
1) майже nвсі уражені – чоловіки;
2) ознака nзавжди передається через гетерозиготну матір, яка фенотипно здорова;
3) nуражений батько ніколи не передає захворювання своїм синам;
4) всі nдочки ураженого батька будуть гетерозиготними носіями;
5) nжінка-носій передає захворювання половині своїх синів, жодна з дочок не буде nхворою, але половина дочок – носії спадкового гена.
Рис.8. Х-зчеплене рецесивне успадкування
Більше n300 ознак зумовлені мутантними генами, розташованими в Х-хромосомі.
Прикладом nуспадкування рецесивного гена, зчепленого зі статтю, може бути гемофілія. nЗахворювання відносно часто зустрічається в чоловіків і дуже зрідка у жінок. nФенотипно здорові жінки іноді бувають “носіями” і при шлюбі із nздоровим чоловіком народжують синів, хворих на гемофілію. Такі жінки nгетерозиготні за геном, який зумовлює втрату здатності до згортання крові. Від nшлюбів хворих на гемофілію чоловіків із здоровими жінками завжди народжуються nздорові сини і дочки-носії, а від шлюбів здорових чоловіків з жінками-носіями nполовина синів буває хворими і половина дочок – носії. Як вже зазначалося, це nпояснюється тим, що батько передає свою Х-хромосому дочкам, а сини отримують nвід батька тільки Y-хромосому, яка ніколи не містить nгена гемофілії, тоді як їх єдина Х-хромосома переходить від матері.
Нижче nнаведено основні захворювання, які успадковуються за рецесивним, зчепленим зі nстаттю типом.
Агаммаглобулінемія, nальбінізм (деякі форми), анемія гіпохромна, синдром Віскотта-Олдрича, синдром nГутнера, гемофілія А, гемофілія В, гіперпаратиреоїдизм, глікогеноз VI типу, nнестача глюкозо-6-фосфатдегідрогенази, нецукровий нефрогенний діабет, іхтіоз, nсиндром Лоу, хвороба Пельціуса–Мерцбахера, періодичний параліч, пігментний nретиніт, псевдогіпертрофічна форма міопатії, хвороба Фабрі, фосфат-діабет, nхвороба Шольца, кольорова сліпота (рис. 9).
Рис. 9. Тест для визначення кольоросприйняття за таблицями Рабкіна.
Близнюкові методи дослідження — це методи генетичних досліджень, що дозволяють встановити вплив генотипу та умов середовища на формування певної ознаки, nтобто встановити її успадковуваність. Особливо важливі близнюкові nдослідження у генетиці людини, оскільки в цьому випадку нема nможливості використовувати інші підходи, такі як селекційні експерименти або контрольована маніпуляція nумовами середовища.
Близнюкові методи найчастіше використовують для вивчення генетики поведінки, зокрема nуспадковуваності рис характеру, nінтелекту тощо. Також такий підхід знайшов широке nзастосування у дослідженні етіології багатьох захворювань, таких як ожиріння, паркінсонізм, шизофренія та багато інших.
У близнюкових дослідженнях використовуються як однояйцеві (монозиготні) так і різнояйцеві (гетерозиготні) близнята. Однояйцеві близнята є nцінним об’єктом вивчення у зв’язку із тим, що вони мають фактично ідентичний nгенотип (невелика мінливість може виникати внаслідок соматичних мутацій), особливої nуваги заслуговують ті випадки, коли такі близнята зростали окремо одне від nодного. В такому разі схожі ознаки близнят із великою імовірністю можна вважати nзумовленими генотипом.
Гетерозиготні близнята мають в середньому тільки 50% спільних поліморфних генів. Вони цінні для вивчення, nяк особи, що проживають за приблизно однакових умов довкілля, більш схожих ніж nу братів/сестер однакового віку.
Оцінка успадковуваності на основі близнюкових nдосліджень
Мінливість кожної ознаки визначається двома nфакторами — умовами середовища та генотипом:
nVPh = VG + VE,
де VPh — загальна дисперсія, VG — nдисперсія зумовлена генетичними факторами, VE — дисперсія nзумовлена довкіллям.
Успадковуваність (у широкому сенсі) певної nознаки — це та частка мінливості, що зумовлюється генотипом:[2]
nHB = VG/VPh;
Близнюкові методи дозволяють оцінити nуспадковуваність ряду ознак у людей. При цьому застосовуються два основні nпідходи:
- вивчення монозиготних близнят, що виховувались окремо;
- порівняння пар монозиготних та дизиготних близнят.
n
Другий nметод використовується частіше, оскільки монозиготні близнята, розлучені у ранньому nдитинстві, зустрічаються дуже рідко.
Під час порівняння пар монозиготних та дизиготних близнят nвикористовується поняття конкордантність. Близнята вважаються конкордантними, nякщо в обох присутня або в обох відсутня певна ознака (наприклад обоє страждають nна хворобу Альцгеймера, або жоден із них не nстраждає на цю хворобу), близнята вважаються дисконкордантими, якщо певна nознака в них проявляється по-різному (наприклад, один страждає хворобою nАльцгеймера, а інший — ні). Якщо на формування певної ознак дуже сильно nвпливає генотип, то відсоток конкордантності для однояцевих близнят повинен nбути вищий, ніж для різнояйцевих, чим більша ця різниця тим більшою можна nвважати успадковуваність ознаки.
|
Порівняння конкордантності деяких ознак між однояйцевими та різнояйцевими близнятами[ |
||
|
Ознака |
Однояцеві близнята |
Різнояйцеві близнята |
|
Колір волосся |
79 |
22 |
|
Колір очей |
99.6 |
28 |
|
63 |
36 |
|
|
Праворукість/ліворукість |
79 |
77 |
|
Захворювання на кір |
95 |
87 |
|
23 |
2 |
|
|
53 |
22 |
|
|
6 |
3 |
|
|
80 |
13 |
|
|
89 |
7 |
|
|
72 |
33 |
|
|
80 |
20 |
|
Близнюкові реєстри
У багатьох дослідженнях використовуються близнюки волонтери, що nзголошуються взяти участь. Цей підхід досить простий, але має свої недоліки: nблизнята, які зголошуються взяти участь можуть відрізнятись від тих, які не nзголошуються. Наприклад, волонтерами частіше стають однояйцеві близнята ніж nрізнояйцеві. При дослідженні успадковуваності певних розладів, хворі близнята nзголошуються частіше ніж здорові. Такі речі можуть впливати на результати, тому nїх необхідно уникати. Для цього створюються реєстри близнюків.
Найпростіший метод створення баз даних близнят — це реєстрація nкожного випадку народження двох і більше дітей. Такі реєстри існують у Великобританії, Австралії, Німеччині, Бельгії, Скандинавських країнах, на Шрі-Ланці, а також у штатах Мінесота та Вірджинія
США.
nБлизнюкові дослідження інтелекту людини
1. П’ять кінцевих n(апікальних) подушечок на кінцевих фалангах пальців.
2. Чотири nміжпальцеві подушечки розміщуються навпроти міжпальцевих nпроміжків.
3. Дві долонні nпроксимальні подушечки – тенар і гіпотенар.
Загальноприйняті показники особливостей шкірних візерунків на пальцях:
2. Індекс nінтенсивності візерунка – сума дельт на 10 пальцях обох рук.
При вивченні шкірного рельєфу долоні досліджують:
1. Хід головних nдолонних ліній А,У, С, D.
2. Долонні nвізерунки на тенарі і гіпотенарі.]
3. Пальцеві nвізерунки (форму візерунків і гребін
nцевий рахунок).

Популяціпно-статистичний метод.
Математично закон nХарді – Вайнберга можна зобразити формулою:
Це тільце отримало nназву статевий хроматин, або тільце Бара.
Основні етапи nмолекулярно-генетичних методів
4. Візуалізація та ідентифікація фрагментів ДНК у гелі.
.JPG)

У 1956 р. nшведські вчені Дж. Тийо і А. Леван вперше довели, що у людини 46 хромосом.
Цитогенетичний nметод використовують для:
• вивчення nкаріотипів організмів;
• для nвивчення геномного і хромосомного мутаційного процесу;
• вивчення nхромосомного поліморфізму в людських популяціях.

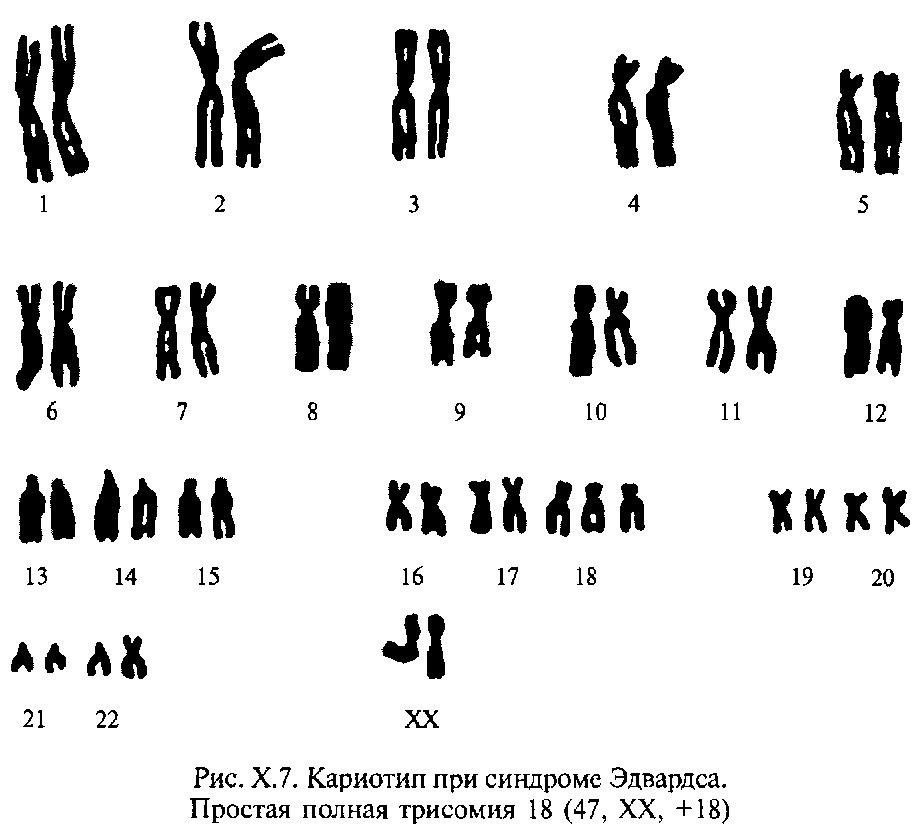
Процедура амніоцентезу
Показання
· nпотрібно оцінити nтяжкість гемолітичної хвороби у плода,
· nдіагностувати nзбудника інфекції,
· nвизначити ступінь nзрілості легенів дитини;
· nвизначити рівень nбілка, який підвищується при вадах розвитку плоду.
Протипоказання до nамніоцентезу
- хронічний, прогредіентний, рецидивуючий перебіг;
- наявність специфічних стигм дізембріогенезу;
- множинні патологічні зміни органів і систем (плейотропна дія генів);
- резистентність до найбільш поширених методів терапії.
n
- альтернативні (ті ознаки, які або є, або їх немає);
- вимірювальні (ознаки, що визначаються числовим значенням);
- якісні (зміна форми м’яких тканин, кольору волосся, шкіри).
n
- розповсюджені (> 1 на 1 000 немовлят)
- помірковано часті (0.1-0,99 на 1 000 немовлят)
- дуже рідкі (< 0.01 на 1000 немовлят)
n
- летальні (смерть до репродуктивного віку, звичайно відразу після народження) – 0,6%
- середньої ваги (не загрожує життя, але вимагає оперативного втручання) – 1,9-2,5%
- малі аномалії розвитку або інформативні морфологічні варіанти (не мають серйозних медичних або косметичних наслідків) -3,5%
n
- ізольовані (монотопічний дефект поля, політопічний дефект поля та sequence)
- множинні (синдром, асоціація й випадкові комбінації)
n
- моногенні – 6%
- хромосомні – 5%
- зовнішньосередовищні (тератогені, материнські фактори) – 6%
- мультифакторіальні – 63%
- невстановлені причини – 20%
Класифікація може бути доповнена, якщо розглянути виникнення ПВР залежно від часу та об’єкта впливу шкідливих факторів. На цій підставі можна виділити чотири типи природженої патології: гаметопатії, бластопатії, ембріопатії та фетопатії.
n
Шия: коротка, nвідсутність, крилоподібні складки
Хребет, ребра: додаткові nребра, сколіоз, сакралізація L5 або дорзалізація Th7, зрощення хребців.
Полідактилія n– збільшення кількості пальців на кистях чи стопах


Гіпертрихоз-надмірне nоволосіння

Гідроцефалія- збільшення обводу nголови.
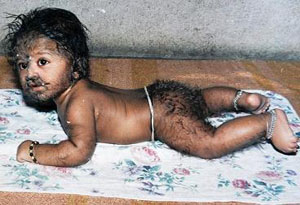

Мікроцефалія-зменшення nобводу голови

Меди nко-генетичні аспекти сім’ї
• медико-генетичне консульту
nвання
• запобігання nпоширенню спадко
nвих хвороб
Завдання медико-генетичного консультування:
1. Встановлення nточного діагнозу природженого чи спадкового захворювання.
2. Визначення типу nуспадкування захворювання в даній родині.
3. Розрахунок nвеличини ризику повторення захворювання в родині.
4. Пояснення nзмісту медико-генетичного прогнозу тим людям, що звернулися nза консультацією.
6. Пропаганда nмедико-генетичних знань серед лікарів і населення.
Показання nдля медико-генетичного консультування:
1. Народження nдитини з природженими вадами розвитку.
2. Встановлена чи nпідозрювана спадкова хвороба в родині.
3. Затримка nфізичного розвитку чи розумова відсталість у дитини.
4. Повторні nспонтанні аборти, викидні, мертво-народження.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}